Рекомендация Коллегии Евразийской экономической комиссии от 27.10.2020 N 18
КОЛЛЕГИЯ ЕВРАЗИЙСКОЙ ЭКОНОМИЧЕСКОЙ КОМИССИИ
РЕКОМЕНДАЦИЯ
от 27 октября 2020 г. N 18
О РУКОВОДСТВЕ
ПО ИССЛЕДОВАНИЮ ФАРМАКОЛОГИЧЕСКОЙ БЕЗОПАСНОСТИ
ЛЕКАРСТВЕННЫХ ПРЕПАРАТОВ ДЛЯ МЕДИЦИНСКОГО ПРИМЕНЕНИЯ
Коллегия Евразийской экономической комиссии в соответствии со статьей 30 Договора о Евразийском экономическом союзе от 29 мая 2014 года, пунктом 3 статьи 3 и статьей 6 Соглашения о единых принципах и правилах обращения лекарственных средств в рамках Евразийского экономического союза от 23 декабря 2014 года, а также в целях гармонизации требований, предъявляемых к исследованию фармакологической безопасности лекарственных препаратов для медицинского применения, установленных законодательством государств - членов Евразийского экономического союза,
рекомендует государствам - членам Евразийского экономического союза по истечении 6 месяцев с даты опубликования настоящей Рекомендации на официальном сайте Евразийского экономического союза при исследовании фармакологической безопасности лекарственных препаратов для медицинского применения использовать Руководство согласно приложению.
Врио Председателя Коллегии
Евразийской экономической комиссии
В.НАЗАРЕНКО
Приложение
к Рекомендации Коллегии
Евразийской экономической комиссии
от 27 октября 2020 г. N 18
РУКОВОДСТВО
ПО ИССЛЕДОВАНИЮ ФАРМАКОЛОГИЧЕСКОЙ БЕЗОПАСНОСТИ
ЛЕКАРСТВЕННЫХ ПРЕПАРАТОВ ДЛЯ МЕДИЦИНСКОГО ПРИМЕНЕНИЯ
I. Общие положения
1. Настоящее Руководство разработано в целях защиты участников клинических исследований (пациентов), получающих зарегистрированные лекарственные препараты, от нежелательного действия лекарственных препаратов, а также в целях сокращения использования лабораторных животных и рационального использования других ресурсов при проведении доклинических и клинических исследований фармакологической безопасности лекарственных препаратов для медицинского применения.
Фармакологические исследования подразделяются на три категории: исследования первичной фармакодинамики, исследования вторичной фармакодинамики и исследования фармакологической безопасности.
2. Настоящее Руководство содержит рекомендации по исследованию фармакологической безопасности лекарственных препаратов для медицинского применения.
3. Настоящее Руководство распространяется на новые химические соединения и биотехнологические лекарственные препараты для медицинского применения. Также настоящее Руководство может применяться в отношении зарегистрированных лекарственных препаратов (например, в связи с выявлением новых нежелательных реакций, изменением популяции пациентов или добавлением нового пути введения лекарственного препарата).
4. При планировании и проведении исследований фармакологической безопасности лекарственных препаратов для медицинского применения следует придерживаться рационального подхода. Вид и дизайн исследований фармакологической безопасности лекарственных препаратов для медицинского применения зависят от свойств и предполагаемого применения лекарственных препаратов. Следует использовать научно обоснованные методы и технологии исследования, предпочтительно признанные в мировой практике.
5. Допускается включать часть конечных точек фармакологической безопасности в дизайн токсикологических, кинетических, клинических и других исследований, однако окончательная оценка фармакологической безопасности проводится только в рамках специальных исследований фармакологической безопасности лекарственных препаратов для медицинского применения, позволяющих обнаружить нежелательные реакции, которые могут быть не выявлены в ходе стандартных токсикологических исследований.
II. Определения
6. Для целей настоящего Руководства используются понятия, которые означают следующее:
"исследования вторичной фармакодинамики" - изучение механизма действия и (или) эффектов исследуемого вещества, не связанных с его целевой терапевтической мишенью, такие исследования иногда рассматриваются как часть исследований общей фармакологии исследуемого вещества;
"исследования первичной фармакодинамики" - изучение механизма действия и (или) эффектов исследуемого вещества связанных с его целевой терапевтической мишенью;
"исследования фармакологической безопасности" - исследования, направленные на изучение потенциальных нежелательных фармакодинамических эффектов исследуемого вещества на физиологические функции организма, при его применении в дозах, соответствующих терапевтическому диапазону и выше.
7. В некоторых случаях информация о первичных и вторичных фармакодинамических эффектах исследуемого вещества может учитываться при проведении оценки безопасности лекарственного препарата и вызываемых им потенциальных нежелательных реакций у человека, поэтому такая информация рассматривается вместе с результатами исследований фармакологической безопасности.
III. Планирование и организация исследований
фармакологической безопасности
8. Целями исследований фармакологической безопасности являются:
выявление нежелательных фармакодинамических свойств исследуемого вещества, которые могут оказать негативное воздействие на здоровье человека;
оценка нежелательных фармакодинамических и (или) патофизиологических эффектов исследуемого вещества, выявленных в ходе токсикологических и (или) клинических исследований;
исследование механизма наблюдаемых и (или) подозреваемых нежелательных фармакодинамических эффектов исследуемого вещества.
В разделе 2.4 модуля 2 регистрационного досье лекарственного препарата следует представить подробный план исследования фармакологической безопасности, направленный на достижение указанных целей.
1. Общие вопросы выбора и планирования исследований
фармакологической безопасности
9. Поскольку фармакологические эффекты зависят от свойств исследуемого вещества, следует выбирать и планировать исследования фармакологической безопасности, с учетом следующих факторов:
эффекты, характерные для фармакологического класса, к которому относится исследуемое вещество, так как они позволяют предположить развитие определенных нежелательных реакций (например, аритмогенная нежелательная реакция является общим свойством антиаритмических лекарственных препаратов);
нежелательные реакции, характерные для химического и фармакологического классов, но не зависящие от первичной фармакодинамики (например, способность антипсихотических лекарственных препаратов удлинять интервал QT);
данные о связывании исследуемого вещества с рецепторами или влиянии на ферментные системы, свидетельствующие о возможности развития нежелательных реакций;
результаты ранее проведенных исследований фармакологической безопасности, вторичной фармакодинамики, токсикологических исследований или клинического применения, требующие дальнейшего изучения, с учетом их значимости для оценки возможных нежелательных реакций у человека.
10. На ранних стадиях разработки лекарственного препарата могут отсутствовать данные, необходимые для выбора и планирования исследования фармакологической безопасности, предусмотренные пунктом 9 настоящего Руководства (например, о сравнительном метаболизме). В таком случае применяются более общие подходы к проведению исследований фармакологической безопасности.
11. Иерархия систем органов может быть разработана в соответствии с их значимостью для жизненно важных функций организма человека. В первую очередь в рамках исследований фармакологической безопасности следует изучить действие лекарственного препарата на жизненно важные органы и системы: сердечно-сосудистую, дыхательную и центральную нервную системы. Остальные системы органов (например, мочевыделительная и пищеварительная системы), функции которых могут временно нарушаться ввиду нежелательных фармакодинамических эффектов без причинения необратимого вреда, не требуют немедленного изучения. В зависимости от таких факторов, как планируемое клиническое исследование или популяция пациентов, может потребоваться оценка фармакологической безопасности влияния лекарственного препарата на функции других органов и систем организма (например, на пищеварительную систему при болезни Крона, функцию почек при первичной ренальной артериальной гипертензии, иммунную систему у иммунокомпрометированных пациентов).
2. Тест-системы
Выбор тест-систем
12. Выбор подходящей животной модели или другой тест-системы обосновывается и осуществляется на основе следующих факторов:
способность животной модели реагировать на фармакодинамические эффекты;
фармакокинетический профиль, вид, порода, пол и возраст животной модели;
восприимчивость, чувствительность и воспроизводимость тест-системы;
доступность ранее полученных данных об исследуемом веществе.
При выборе тест-системы следует учитывать (при наличии) данные, полученные у человека (например, о метаболизме in vitro). Сроки осуществления измерений должны определяться фармакодинамическими и фармакокинетическими свойствами исследуемого вещества. В целях получения достоверных научных данных необходимо обосновать выбор конкретной животной модели или тест-системы.
Исследования фармакологической безопасности in vitro
и in vivo
13. В качестве тест-систем допускается использовать животные модели, а также системы ex vivo и in vitro, в том числе изолированные органы и ткани, клеточные культуры, клеточные фрагменты, органеллы клеток, рецепторы, ионные каналы, переносчики, ферменты.
Системы in vitro допускается использовать во вспомогательных исследованиях (например, для установления профиля активности исследуемого вещества или для исследования механизма эффектов, наблюдаемых in vivo).
14. При проведении исследований in vivo целесообразно использовать животных, в отношении которых не выполнялась анестезия. Предпочтительно использовать данные, полученные от необездвиженных животных, которые можно регистрировать в течение длительного периода методом телеметрии или другими подходящими методами, предназначенными для животных, находящихся в сознании, или животных, приспособленных к лабораторным условиям, а не данные, полученные от обездвиженных или неприспособленных к лабораторным условиям животных. Одним из главных условий при использовании животных, в отношении которых не выполнялась анестезия, является предотвращение развития у животных дискомфорта и боли.
3. Дизайн исследования фармакологической безопасности
Размер выборки и использование контролей
15. Количество животных или препаратов изолированных органов должно быть достаточным, чтобы подтвердить или исключить наличие биологически значимого эффекта исследуемого вещества, провести полноценную научную интерпретацию полученных данных. При этом следует учитывать величину того биологического эффекта, который потенциально наиболее вероятно проявится у человека. В дизайн исследования фармакологической безопасности следует включить достаточное количество отрицательных и положительных контрольных групп. Для хорошо охарактеризованных тест-систем in vivo положительные контрольные группы могут не понадобиться. Исключение контрольных групп из исследования следует обосновать.
Путь введения исследуемого вещества
16. При проведении исследования целесообразно использовать клинический путь введения исследуемого вещества, предусмотренный проектом его общей характеристики лекарственного препарата. Независимо от пути введения экспозиция исследуемого вещества или его основных метаболитов должна быть сопоставима со значениями, достигаемыми у человека (при наличии таких данных), или превышать их. Если исследуемое вещество предназначено для введения несколькими путями (например, внутрь и парентерально) или если наблюдаются либо ожидаются значительные качественные и количественные различия между системной и местной экспозицией, оценку эффекта исследуемого вещества следует проводить для более чем одного пути его введения.
4. Величины доз или концентрации исследуемого вещества
Исследования фармакологической безопасности in vivo
17. Исследования фармакологической безопасности in vivo следует планировать таким образом, чтобы установить зависимость "доза - ответ" для наблюдаемых нежелательных реакций. По возможности следует изучить зависимость нежелательных реакций от времени после введения исследуемого вещества (например, начало и длительность эффекта). Как правило, следует сравнить дозы, вызывающие нежелательные реакции, с дозами, вызывающими первичный фармакодинамический эффект у исследуемых видов животных или предполагаемый терапевтический эффект у человека (если это возможно). Ввиду наличия межвидовых различий по фармакодинамической чувствительности, дозы исследуемого вещества должны включать и превышать первичный фармакодинамический и терапевтический диапазоны. При отсутствии в ходе исследования нежелательных реакций по изучаемым показателям фармакологической безопасности в качестве высшей исследуемой дозы необходимо выбрать такую дозу, которая вызывает нежелательные реакции средней тяжести в других исследованиях с аналогичным путем введения эквивалентных по длительности. Такие нежелательные реакции могут включать в себя дозолимитирующие фармакодинамические эффекты и другие токсические реакции. При некоторых эффектах, возникающих в диапазоне токсического действия исследуемого вещества, в том числе треморе и фасцикуляции во время снятия электрокардиограммы, может искажается интерпретация результатов, а также требуется уменьшение величины дозы исследуемого вещества. Исследование одной группы лабораторных животных при использовании такой ограничивающей дозы лекарственного препарата может быть достаточным, если нежелательные реакции по конечным точкам фармакологической безопасности у экспериментальных видов животных отсутствуют.
Исследования фармакологической безопасности in vitro
18. Исследования фармакологической безопасности in vitro следует планировать таким образом, чтобы установить зависимость "концентрация - ответ" для исследуемого вещества. Необходимо выбрать диапазон концентраций исследуемого вещества, позволяющий увеличить вероятность выявления ответной реакции используемой тест-системы. На верхнюю границу такого диапазона могут повлиять физико-химические свойства исследуемого вещества и другие специфичные для метода факторы. При отсутствии эффекта следует обосновать выбранный диапазон концентраций исследуемого вещества.
5. Длительность исследований фармакологической безопасности
19. Исследования фармакологической безопасности, как правило, предусматривают однократное введение исследуемого вещества. В случае проявления нежелательных фармакодинамических эффектов в процессе лечения или возникновения опасения в отношении его фармакологической безопасности по итогам доклинических исследований с повторным (многократным) введением исследуемого вещества либо в результате применения исследуемого вещества у человека, необходимо выбрать длительность исследований фармакологической безопасности таким образом, чтобы учесть возможность установления фармакодинамических эффектов экспериментальным путем.
6. Изучение метаболитов, изомеров и готовых
лекарственных препаратов
20. В исследованиях фармакологической безопасности, как правило, изучается каждое исходное вещество и его основные метаболиты, достигающие или способные достичь системной экспозиции у человека. Оценка основных метаболитов часто осуществляется в рамках исследования на животных. Если выясняется, что основные метаболиты у человека отсутствуют, а у животных образуются только в относительно низких концентрациях, следует изучить влияние таких метаболитов на конечные точки фармакологической безопасности. Кроме того, в случае если метаболиты человека оказывают существенное влияние на фармакологическое действие лекарственного препарата, такие метаболиты следует изучить. Если в исследованиях in vivo исходного соединения метаболиты должным образом не изучались допускается изучать метаболиты в системах in vitro.
21. Если лекарственный препарат представляет собой смесь изомеров исследуемого вещества, следует изучить каждый отдельный изомер in vitro или in vivo.
22. Исследования фармакологической безопасности готового лекарственного препарата проводятся только в случае, если производственная рецептура и (или) специфика производства готовой лекарственной формы изменяют фармакокинетику и (или) фармакодинамику действующего вещества лекарственного препарата по сравнению с ранее изученными производственной рецептурой и (или) спецификой производства готовой лекарственной формы (то есть за счет таких активных вспомогательных веществ, как усилители проникающей способности, липосомы и другие изменения, например полиморфизм).
7. Основная батарея исследований
фармакологической безопасности
23. Целью основной батареи исследований фармакологической безопасности является изучение влияния исследуемого вещества на жизненно важные функции организма человека. В связи с этим сердечно-сосудистая, дыхательная и центральная нервная системы, как правило, считаются витальными системами органов, требующими изучения в рамках основной батареи исследований фармакологической безопасности. В некоторых случаях, на основании научных данных, основная батарея исследований фармакологической безопасности должна включать в себя дополнительные исследования, указанные в подразделе 9 настоящего раздела. При наличии условий, приведенных в подразделе 10 настоящего раздела, основная батарея исследований фармакологической безопасности может не проводиться.
24. Исключение из основной батареи исследований фармакологической безопасности отдельного теста (тестов) или исследования определенных органов, систем или функций организма должно быть обосновано научными данными.
25. Следует должным образом изучить влияние исследуемого вещества на:
центральную нервную систему (оценить двигательную активность, изменение поведения, координацию движений, сенсорные рефлексы (моторные рефлексы) и температуру тела у тест-системы (субъекта исследования)) с использованием батареи тестов функционального наблюдения, модифицированного теста Ирвина или других тестов;
сердечно-сосудистую систему (оценить артериальное давление, частоту сердечных сокращений и электрокардиограмму, а также изучить данные, полученные in vivo, in vitro и (или) ex vivo, включая методы оценки нарушений реполяризации и проведения в миокарде, согласно приложению к настоящему Руководству);
дыхательную систему (оценить частоту дыхания и другие параметры функции дыхания (например, дыхательный объем или насыщение гемоглобина кислородом)). Для оценки функции дыхания клинического наблюдения за лабораторными животными может оказаться недостаточно, поэтому следует проводить количественное измерение указанных параметров с использованием подходящей методологии методики.
8. Последующие и дополнительные исследования
фармакологической безопасности
26. Основываясь на фармакологических свойствах и химическом классе исследуемого вещества, можно предположить возможность развития нежелательных реакций. Кроме того, по результатам проведения основной батареи исследований фармакологической безопасности, клинических исследований, фармаконадзора, экспериментальных исследований in vitro и in vivo или на основе научных данных могут возникать дополнительные опасения относительно безопасности лекарственного препарата.
27. В случае возникновения таких опасений или развития нежелательных реакций их следует должным образом изучить в рамках последующих или дополнительных исследований фармакологической безопасности.
28. В пунктах 30 - 37 настоящего Руководства приводится перечень исследований фармакологической безопасности, направленных на дальнейшее изучение указанных систем органов для выявления потенциальных нежелательных фармакодинамических эффектов. Данный перечень исследований не является полным или обязательным. В некоторых случаях нежелательные реакции целесообразно изучать в рамках других доклинических и (или) клинических исследований, которые следует подбирать в индивидуальном порядке с учетом ранее полученных доклинических и клинических данных.
Последующие исследования в рамках основной батареи
исследований фармакологической безопасности
29. Последующие исследования основной батареи исследований фармакологической безопасности в целях подтверждения результатов основной батареи исследований жизненно важных функций организма или получения дополнительных данных.
30. В рамках исследования центральной нервной системы следует изучить поведенческую фармакологию, способность к обучению и функцию памяти, лиганд-специфическое связывание, нейрохимию, зрительные, слуховые функции и (или) провести электрофизиологические исследования и т.д.
31. В рамках исследования сердечно-сосудистой системы следует изучить сердечный выброс, сократимость желудочков, сопротивляемость сосудов, влияние эндогенных и (или) экзогенных веществ на сердечно-сосудистую систему и т.д.
32. В рамках исследования дыхательной системы следует изучить сопротивление дыхательных путей, эластичность легочной ткани, легочное артериальное давление, газы крови, pH крови и т.д.
Дополнительные исследования фармакологической безопасности
33. Дополнительные исследования фармакологической безопасности проводятся для оценки потенциальных нежелательных фармакодинамических эффектов со стороны функций систем органов, не изученных в рамках основной батареи исследований или исследований токсичности с повторным (многократным) введением исследуемого вещества, при наличии основания для проведения такой оценки.
34. В рамках исследования мочевыделительной системы следует изучить влияние исследуемого вещества на показатели работы почек (исследовать объем и плотность мочи, осмоляльность, pH, водно-электролитный баланс, содержание белка в моче, цитологию мочи, а также показатели биохимии крови (азот мочевины крови, креатинин и белки плазмы)).
35. В рамках исследования автономной нервной системы следует изучить влияние исследуемого вещества на автономную нервную систему (связывание исследуемого вещества с рецепторами автономной нервной системы, функциональную реакцию на применение агонистов или антагонистов in vitro или in vivo, прямую стимуляцию автономных нервов, и реакцию сердечно-сосудистой системы, оценить барорефлексы, вариацию сердечного ритма).
36. В рамках исследования пищеварительной системы следует изучить влияние исследуемого вещества на пищеварительную систему (например, исследовать желудочную секрецию, потенциал поражения желудочно-кишечного тракта, секрецию желчи, длительность транзита in vivo, сократимость подвздошной кишки in vitro, измерить значения pH желудка и задержки пищи в желудке).
37. При наличии оснований следует оценить влияние исследуемого вещества на другие системы органов, не изученные ранее (например, провести исследования потенциала развития лекарственной зависимости, мышечной, иммунной и эндокринной функций).
9. Условия, при которых исследования фармакологической
безопасности не проводятся
38. Если фармакологические свойства исследуемого вещества для местного применения хорошо охарактеризованы и установлена его низкая системная экспозиция или распределение в другие органы и ткани, то исследования фармакологической безопасности могут не проводиться.
39. Исследования фармакологической безопасности исследуемого вещества до первого введения лекарственного препарата человеку могут не потребоваться для цитотоксических лекарственных препаратов, предназначенных для лечения пациентов с терминальными формами рака. Однако результаты исследований фармакологической безопасности цитотоксических лекарственных препаратов с новым механизмом действия могут представлять ценность для последующей оценки профиля безопасности исследуемого вещества и родственных ему соединений.
40. Для биотехнологических лекарственных препаратов с высокой специфичностью в отношении рецептора-мишени достаточно оценить конечные точки фармакологической безопасности в рамках токсикологических и (или) фармакодинамических исследований, поэтому программа исследований фармакологической безопасности таких лекарственных препаратов может быть сокращена или исключена.
41. Для биотехнологических лекарственных препаратов, представляющих собой новый терапевтический класс, и (или) подобных лекарственных препаратов, не обладающих высокой специфичностью в отношении рецептора-мишени, следует предусмотреть более детальные исследования фармакологической безопасности.
42. Возможны другие исключения, не требующие проведения исследований фармакологической безопасности (например, новая соль, обладающая теми же фармакокинетическими и фармакодинамическими свойствами, что и ранее изученная соль исследуемого вещества).
10. Сроки проведения исследований
фармакологической безопасности по отношению к программе
клинической разработки лекарственного препарата
43. При планировании программы изучения фармакологической безопасности следует учитывать условия, указанные в пунктах 38 - 42 настоящего Руководства, чтобы определить необходимость выполнения определенных исследований.
Исследования, которые следует провести до первого введения
исследуемого вещества человеку
44. Перед первым введением исследуемого вещества человеку следует изучить влияние исследуемого вещества на жизненно важные функции организма человека, перечисленные в основной батарее исследований фармакологической безопасности. Также следует провести все необходимые последующие и дополнительные исследования. Данные хорошо спланированных и проведенных токсикологических исследований, в рамках которых изучались конечные точки фармакологической безопасности, могут быть использованы для сокращения или исключения необходимости проведения отдельных исследований фармакологической безопасности.
Исследования, проводимые в ходе программы клинической
разработки лекарственного препарата
45. В целях углубленного изучения наблюдавшихся или подозреваемых нежелательных реакций у животных или человека в ходе клинической разработки может потребоваться проведение дополнительных исследований фармакологической безопасности.
Исследования фармакологической безопасности, проводимые
до регистрации лекарственного препарата
46. До регистрации лекарственного препарата следует оценить его влияние на системы органов в соответствии с пунктами 26 - 37 настоящего Руководства. Отсутствие необходимости проведения исследований фармакологической безопасности до регистрации лекарственного препарата следует обосновать. Доступные данные хорошо спланированных и проведенных токсикологических исследований, в рамках которых изучались конечные точки фармакологической безопасности, и данные клинических исследований могут способствовать такой оценке и заменить собой исследования фармакологической безопасности.
11. Соответствие исследований Правилам надлежащей
лабораторной практики Евразийского экономического союза
47. Доклинические исследования фармакологической безопасности, как правило, должны проводиться в соответствии с Правилами надлежащей лабораторной практики Евразийского экономического союза в сфере обращения лекарственных средств, утвержденными Решением Совета Евразийской экономической комиссии от 3 ноября 2016 г. N 81 (далее - Правила надлежащей лабораторной практики). Однако вследствие уникального дизайна и практических особенностей некоторых исследований фармакологической безопасности обеспечить их соответствие Правилам надлежащей лабораторной практики не представляется возможным.
В случае если исследования фармакологической безопасности не соответствуют Правилам надлежащей лабораторной практики, необходимо обеспечить высокое качество получаемых данных и целостность всех исследований, а также возможность воссоздания исследований путем надлежащего ведения документации и архивирования данных. Необходимость проведения любого исследования (или его части), не соответствующее Правилам надлежащей лабораторной практики, следует должным образом обосновать и описать в отчете об исследовании потенциального влияния дизайна и методологии исследования на оценку конечных точек фармакологической безопасности.
48. Основная батарея исследований фармакологической безопасности, как правило, проводится в соответствии с Правилами надлежащей лабораторной практики. Последующие и дополнительные исследования должны (насколько это возможно) соответствовать Правилам надлежащей лабораторной практики. В случае если исследование фармакологической безопасности проводится в составе токсикологических исследований, последние следует проводить в соответствии с Правилами надлежащей лабораторной практики.
49. Исследования первичной фармакодинамики не требуется проводить в соответствии с Правилами надлежащей лабораторной практики. Исследования вторичной фармакодинамики, как правило, могут не соответствовать Правилам надлежащей лабораторной практики. Результаты исследований вторичной фармакодинамики, полученные в ходе выбора соединения, могут учитываться при проведении оценки фармакологической безопасности.
В случаях если результаты исследований вторичной фармакодинамики оказывают существенное влияние на оценку фармакологической безопасности потенциальных нежелательных реакций у человека, такие исследования проводят в соответствии с Правилами надлежащей лабораторной практики.
В случае если отсутствуют основания для отрицательной оценки качества проведенных исследований (например, конечные точки фармакологической безопасности не достигнуты, результаты по химическому или терапевтическому классу исследуемого вещества не достигнуты), отсутствует необходимость повторно проводить исследования в соответствии с Правилами надлежащей лабораторной практики.
Приложение
к Руководству по исследованию
фармакологической безопасности
лекарственных препаратов
для медицинского применения
УКАЗАНИЯ
ПО ДОКЛИНИЧЕСКОЙ ОЦЕНКЕ СПОСОБНОСТИ ИССЛЕДУЕМОГО ВЕЩЕСТВА
ВЫЗЫВАТЬ ЗАМЕДЛЕНИЕ РЕПОЛЯРИЗАЦИИ ЖЕЛУДОЧКОВ СЕРДЦА
(УДЛИНЯТЬ ИНТЕРВАЛ QT)
I. Общие положения
1. Оценка влияния лекарственных препаратов на реполяризацию желудочков сердца и аритмогенный риск является предметом активного изучения на доклиническом и клиническом этапе разработки лекарственных препаратов.
2. Настоящие Указания описывают стратегию доклинических исследований оценки потенциала исследуемого действующего вещества замедлять реполяризацию желудочков сердца и содержат сведения о доклинических анализах и интегральной оценке риска.
3. Интервал QT (время от начала комплекса QRS до конца зубца T) на электрокардиограмме (ЭКГ) - мера длительности деполяризации и реполяризации желудочков сердца. Удлинение интервала QT может быть врожденным или приобретенным (например, лекарственно обусловленным). При замедлении реполяризации желудочков сердца и удлинении интервала QT повышается риск желудочковых тахиаритмий, включая пируэтную желудочковую тахикардию, особенно в сочетании с другими факторами риска (например, гипокалиемией, органическими заболеваниями сердца, брадикардией). В связи с этим уделяется большое внимание потенциальным аритмогенным эффектам лекарственных препаратов, связанных с удлинением интервала QT.
4. Реполяризация желудочков сердца, определяемая по длительности потенциала действия миокарда, - сложный физиологический процесс и совокупный результат работы многих мембранных ионных каналов и переносчиков. В физиологических условиях функции указанных ионных каналов и переносчиков сильно взаимозависимы. На активность каждого ионного канала или переносчика влияет множество факторов, включая внутриклеточную и внеклеточную концентрацию ионов, мембранный потенциал, межклеточное электрическое взаимодействие, ритм сердца, активность автономной нервной системы и т.п. Также важны метаболическое состояние (например, кислотно-основный баланс), расположение и вид кардиомиоцита. Потенциал действия желудочков сердца состоит из 5 последовательных фаз:
фаза 0: нарастание потенциала действия главным образом обусловлено быстрым транзиторным входящим током Na+ (INa) через Na+-каналы;
фаза 1: прекращение нарастания потенциала действия и фаза ранней реполяризации происходят за счет инактивации Na+-каналов и транзиторного выходящего тока K+ (Ito) через K+-каналы;
фаза 2: плато потенциала действия отражает баланс между входящим током Ca2+ (ICa) через Ca2+-каналы L-типа и выходящими реполяризующими K+-токами;
фаза 3: устойчивый нисходящий ход потенциала действия и фаза поздней реполяризации происходят за счет выходящего тока K+ (IKr и IKs) через K+-каналы задержанного выпрямления;
фаза 4: потенциал покоя поддерживается за счет входящего выпрямляющего K+-тока (IKi).
5. Удлинение потенциала действия может происходить из-за сниженной инактивации входящих Na+- или Ca2+-токов, повышенной активации Ca2+-тока или ингибирования одного либо нескольких выходящих K+-токов. Быстро и медленно активирующиеся компоненты K+-тока задержанного выпрямления (IKr и IKs), видимо, играют наиболее важную роль в определении продолжительности потенциала действия и следовательно интервала QT. Ген hERG (human ether-a-go-go-related gene) и ген KvLQT1 кодируют порообразующие белки KCNH2 и KCNQ1, которые, предположительно, представляют ![]() -субъединицу калиевых каналов, отвечающих за IKr и IKs соответственно. Указанные
-субъединицу калиевых каналов, отвечающих за IKr и IKs соответственно. Указанные ![]() -субъединичные белки могут образовывать гетеро-олигомерные комплексы со вспомогательными
-субъединичные белки могут образовывать гетеро-олигомерные комплексы со вспомогательными ![]() -субъединицами (то есть продуктами генов MiRP и MinK), которые, видимо, модулируют пропускные свойства белков-каналов. Наиболее распространенным механизмом удлинения интервала QT под влиянием лекарственных препаратов является ингибирование калиевого тока задержанного выпрямления, отвечающего за IKr.
-субъединицами (то есть продуктами генов MiRP и MinK), которые, видимо, модулируют пропускные свойства белков-каналов. Наиболее распространенным механизмом удлинения интервала QT под влиянием лекарственных препаратов является ингибирование калиевого тока задержанного выпрямления, отвечающего за IKr.
6. Настоящие Указания применяются в отношении новых химических соединений для медицинского применения и зарегистрированных лекарственных препаратов (например, если нежелательные клинические явления, новая популяция пациентов или новый путь введения вызывают риск развития нежелательного эффекта, не изученного ранее). Условия, при которых исследования не требуются, описаны в настоящем Руководстве.
7. In vitro IKr-анализ и in vivo QT-анализ, описанные в пунктах 16 и 17 настоящих Указаний, проводимые в целях регистрации лекарственных препаратов, следует проводить в соответствии с Правилами надлежащей лабораторной практики Евразийского экономического союза в сфере обращения лекарственных средств, утвержденными Решением Совета Евразийской экономической комиссии от 3 ноября 2016 г. N 81 (далее - Правила надлежащей лабораторной практики). Последующие исследования, описанные в пунктах 20 - 22 настоящих Указаний, должны максимально соответствовать указанным Правилам.
8. Поскольку анализы in vitro и in vivo являются комплементарными подходами, следует проводить оба вида анализа.
9. Экспериментальный подход и признаки риска необходимо индивидуализировать под исследуемое вещество в зависимости от его профилей фармакодинамики, фармакокинетики и безопасности.
II. Планирование и организация исследований влияния
изучаемого вещества на реполяризацию желудочков сердца
10. Цели исследований включают в себя:
а) установление способности исследуемого вещества и его метаболитов замедлять реполяризацию желудочков на тест-системе;
б) соотнесение степени замедления реполяризации желудочков с концентрациями исследуемого вещества и его метаболитов.
11. Результаты исследования следует использовать для:
а) выяснения механизма действия исследуемого вещества;
б) оценки риска замедления реполяризации желудочков и удлинения интервала QT у человека при рассмотрении вместе с другими сведениями.
Принципы выбора и планирования исследований
фармакологической безопасности лекарственных препаратов
для медицинского применения
12. Доклиническая методология позволяет изучить следующие функциональные уровни реализации фармакологического эффекта лекарственного препарата:
ионные токи, измеряемые в изолированных кардиомиоцитах животных или человека, культурах клеточных линий сердца и гетерологичных экспрессирующих системах клонированных ионных каналов человека;
параметры потенциала действия в изолированных препаратах сердца или определенные электрофизиологические параметры, характеризующие продолжительность потенциала действия у анестезированных животных;
параметры электрокардиограммы животных, находящихся в сознании или анестезированных;
аритмогенные эффекты, возникающие в изолированных препаратах сердца или у животных.
Указанные функциональные уровни можно изучать, используя методы in vitro и (или) in vivo. Данные, полученные при изучении таких функциональных уровней, могут быть полезны для прогнозирования аритмогенного эффекта лекарственного препарата у человека.
13. Электрофизиологические исследования in vitro позволяют изучить потенциальные клеточные механизмы, которые нельзя спрогнозировать на основании данных, полученных в исследованиях in vivo. Изменения других сердечно-сосудистых параметров или влияние исследуемого вещества на несколько ионных каналов могут осложнять интерпретацию данных. Эту проблему можно решить с помощью комплементарных оценок, полученных на других системах. Несмотря на то что замедление реполяризации может происходить за счет модуляции ионных каналов нескольких видов, ингибирование IKr - наиболее частый механизм реализации индуцируемого лекарством удлинения интервала QT у человека.
14. Модели in vivo, обладающие полным набором молекулярных, биохимических и физиологических систем, также могут быть информативны с точки зрения ответа человека на исследуемое вещество. Тщательно спланированные и проведенные исследования in vivo позволяют оценить исходное вещество и метаболиты, а также позволяют оценить запас их безопасности в отношения повышения дозы или изменения режима дозирования. Оценка ЭКГ in vivo позволяет получить сведения о свойствах проведения импульсов и внесердечных влияниях различных факторов на проводимость (например, тонусе автономной нервной системы). Исследования параметров потенциала действия позволяют получить сведения об интегральной активности нескольких ионных каналов сердца.
Стратегия доклинических исследований лекарственных
препаратов для медицинского применения
15. Общая стратегия доклинических исследований фармакологической безопасности для оценки риска замедления реполяризации желудочков сердца и удлинения интервала QT представлена на рисунке.
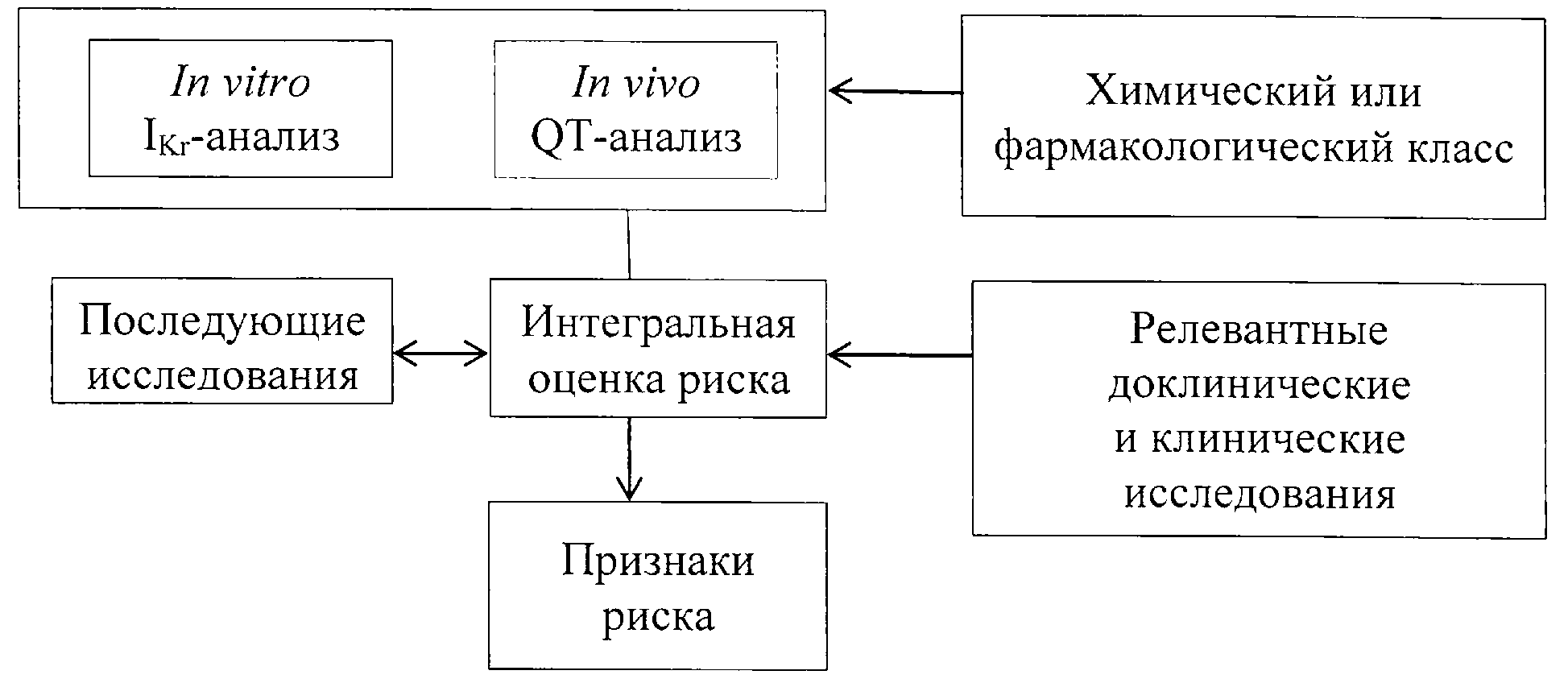
Рис. Стратегия доклинических исследований фармакологической
безопасности лекарственных препаратов
Изучение in vitro IKr-анализа
16. In vitro IKr-анализ оценивает влияние на ионный ток через нативный или экспрессирущий IKr-канальный белок, например, кодируемый геном hERG (пункты 29 - 34 настоящих Указаний).
In vivo QT-анализ
17. In vivo QT-анализ измеряет такие показатели реполяризации желудочков сердца, как интервал QT. Указанный анализ можно спланировать так, чтобы достичь целей как основной батареи исследований фармакологической безопасности (сердечно-сосудистое исследование из основной батареи), так и исследования аритмогенного действия лекарственного препарата в соответствии с настоящими Указаниями. Это позволит снизить использование животных в эксперименте и других ресурсов.
Химический или фармакологический класс
исследуемого вещества
18. Необходимо учесть возможную принадлежность исследуемого вещества к химическому или фармакологическому классу, некоторые члены которого проявляли способность удлинять интервал QT у человека (например, нейролептики, блокаторы H1-гистаминовых рецепторов, фторхинолоны). Указанный фактор следует учитывать (если значимо) при выборе референтного соединения (соединений) и включать в интегральную оценку риска.
Релевантные доклинические и клинические сведения
19. Дополнительные сведения, необходимые для интегральной оценки риска, включают в себя результаты:
а) фармакодинамических исследований;
б) токсикологических исследований или исследований безопасности;
в) фармакокинетических исследований, включая плазменные концентрации исходного вещества и метаболитов (в том числе данные человека (при наличии));
г) исследований лекарственных взаимодействий;
д) исследований распределения и кумуляции в тканях;
е) пострегистрационного изучения.
Последующие исследования
20. Последующие исследования направлены на получение более глубокого понимания или дополнительных знаний о способности исследуемого вещества замедлять реполяризацию желудочков сердца и удлинять интервал QT у человека. Подобные исследования могут позволить получить дополнительные сведения об активности, механизме действия, наклоне кривой "доза - ответ" или величине ответа. Последующие исследования направлены на решение конкретных задач, поэтому применимы различные дизайны исследований in vivo и in vitro.
21. Если результаты разных доклинических исследований не согласованы и (или) результаты клинических исследований отличаются от результатов доклинических исследований, ретроспективная оценка и последующие доклинические исследования могут установить причины возникших расхождений. Результаты последующих исследований фармакологической безопасности могут являться существенным компонентом интегральной оценки риска.
22. При выборе и планировании последующих исследований фармакологической безопасности наряду с релевантными доклиническими и клиническими сведениями необходимо учитывать следующее:
а) выполнение анализов реполяризации желудочков сердца, измеряющих параметры потенциала действия на изолированных препаратах сердца (пункты 29 - 34 настоящих Указаний);
б) использование специфичных электрофизиологических параметров, характеризующих продолжительность потенциала действия у анестезированных животных (пункты 35 - 40 настоящих Указаний);
в) повторное (многократное) введение исследуемого вещества;
г) выбор вида и пола животных;
д) использование индукторов и ингибиторов метаболизма;
е) использование сопутствующих положительных контрольных веществ и референтных соединений (пункты 27 - 28 настоящих Указаний);
ж) ингибирование других ранее не оцененных каналов;
з) измерение электрофизиологических параметров в нескольких временных точках;
и) искажающие эффекты у животных, в отношении которых не применялась анестезия, ограничивающие интерпретацию данных, таких как индуцированные исследуемым веществом влияния на ритм сердца или автономный тонус, либо такой токсичности, как тремор, судороги или рвота.
Интегральная оценка риска
23. Интегральная оценка риска - оценка результатов доклинических исследований, включающая в себя результаты последующих исследований и другие релевантные сведения. Интегральная оценка риска должна быть научной и индивидуализированной под исследуемое вещество. Такая оценка может помогать планированию клинических исследований и интерпретации их результатов. Интегральную оценку (при наличии) риска следует включить в брошюру исследователя и доклинический обзор модуля 2 регистрационного досье лекарственного препарата. В зависимости от стадии разработки лекарственного препарата интегральная оценка риска должна также учитывать:
а) чувствительность и специфичность анализа;
б) активность исследуемого вещества по отношению к референтному соединению (соединениям);
в) зависимость между экспозициями, связанными с влиянием на реполяризацию, и экспозициями исследуемого вещества, оказывающими первичное фармакодинамическое действие в доклинических исследованиях на экспериментальные виды животных или предполагаемое терапевтическое действие на человека;
г) вклад метаболитов в удлинение интервала QT, а также метаболические различия между человеком и животными.
Признаки риска аритмогенного действия
24. Признаки риска аритмогенного действия устанавливаются на основе общего вывода по результатам интегральной оценки риска о способности исследуемого вещества замедлять реполяризацию желудочков сердца и удлинять интервал QT у человека.
Сроки доклинических исследований и интегральной
оценки риска по отношению к программе клинической
разработки лекарственного препарата
25. Следует предусмотреть проведение доклинических исследований, направленных на оценку риска замедления реполяризации желудочков сердца и удлинения интервала QT, перед первым введением исследуемого вещества человеку. Их результаты в составе интегральной оценки риска могут обосновывать планирование и интерпретацию результатов последующих клинических исследований фармакологической безопасности.
III. Тест-системы
Выбор тест-систем
26. Настоящий раздел содержит обзор методологий, используемых для оценки потенциала исследуемого вещества замедлять реполяризацию желудочков сердца и удлинять интервал QT. При выборе наиболее подходящих тест-систем необходимо принимать во внимание:
а) научную достоверность и устойчивость методологии анализа и экспериментальных конечных точек;
б) стандартизацию анализов и лекарственных препаратов;
в) воспроизводимость результатов анализов;
г) релевантность конечных точек или параметров анализов для оценки риска для человека.
Использование положительных контрольных веществ
и референтных соединений
27. В целях демонстрации ответной реакции in vitro препаратов ионных каналов и анализов продолжительности потенциала действия необходимо использовать субмаксимальную эффективную концентрацию положительного контрольного вещества и включать его в каждое исследование. В исследовании in vivo положительные контрольные вещества необходимо использовать для валидации и определения чувствительности тест-системы, но включать их в каждое исследование не требуется.
28. В отношении исследуемых веществ, принадлежащих к химическому или фармакологическому классу, связанному с удлинением интервала QT у человека, следует предусмотреть использование в исследованиях in vitro и in vivo сопутствующего референтного соединения (соединений) (относящегося к тому же химико-фармакологическому классу веществ) в целях облегчения ранжирования активности исследуемого вещества по отношению к его компараторам.
Электрофизиологические исследования in vitro
29. Электрофизиологические исследования in vitro могут позволить получить ценные сведения о влиянии исследуемого вещества на продолжительность потенциала действия и (или) ионные токи в сердце. Эти исследования играют важную роль при оценке потенциала удлинения интервала QT и выяснении клеточных механизмов, влияющих на реполяризацию миокарда. В электрофизиологических исследованиях in vitro используются отдельные клетки (например, гетерологичные экспрессирующие системы, дезагрегированные кардиомиоциты) либо многоклеточные препараты (например, волокно Пуркинье, сосочковая мышца, трабекулы, перфузируемый миокард, интактное сердце). Гетерологичные экспрессирующие системы (белки ионных каналов человека экспрессируются на несердечных клеточных линиях) используются для оценки влияния исследуемого вещества на определенный ионный канал. По сравнению с экспрессирующими системами дезагрегированные миоциты вызывают больше технических затруднений, но позволяют оценить влияние на продолжительность потенциала действия и ионные токи. Несмотря на то что одноклеточные препараты более хрупкие, в них минимизированы диффузионные барьеры в месте действия. Многоклеточные препараты являются стабильными тест-системами для изучения продолжительности потенциала действия. Анализ таких параметров каждой фазы потенциала действия, как Vmax для фазы 0 (INa), APD30 или APD40 для фазы 2 (ICa) и "триангуляция" для фазы 3 (IK), позволяет изучить влияние исследуемого вещества на определенные каналы, отвечающие за эти фазы. Кроме того, некоторые параметры потенциала действия, получаемые с помощью препарата Лангендорффа, позволяют получить сведения об аритмогенном риске.
30. Препараты тканей и клеток для анализов in vitro получают от различных видов лабораторных животных, включая кроликов, хорьков, морских свинок, собак, свиней, а иногда и от человека. Ионные механизмы реполяризации у взрослых крыс и мышей отличаются от ионных механизмов более крупных видов животных, а также человека (основные ионные токи, контролирующие реполяризацию у взрослых крыс и мышей, - Ito), поэтому использование тканей этих видов животных неприемлемо. При выборе тест-системы следует учитывать межвидовые различия с точки зрения того, какие ионные каналы сердца вносят вклад в реполяризацию сердца и продолжительность потенциала действия. При использовании нативных тканей или клеток сердца следует учитывать свойства и источник получения лекарственного препарата, поскольку распределение ионных каналов сердца зависит от расположения и вида клеток.
31. Количество изучаемых концентраций исследуемого вещества в исследованиях in vitro должно охватывать широкий диапазон, покрывающий пределы ожидаемой максимальной терапевтической плазменной концентрации и выходящий за их границы. Возрастающие концентрации исследуются до установления характеристик кривой "концентрация - ответ" или пока физико-химические эффекты не начнут ограничивать концентрацию. В идеале продолжительность экспозиции должна быть достаточной для получения равновесных электрофизиологических эффектов, если этому не препятствует жизнеспособность препарата клеток или тканей. Следует указать продолжительность экспозиции. Для определения чувствительности систем анализа in vitro следует использовать соответствующие положительные контрольные вещества.
32. К факторам, способным искажать или ограничивать интерпретацию результатов электрофизиологических исследований in vitro, относятся:
а) испытанию высоких концентраций исследуемого вещества может препятствовать его низкая растворимость в водных физиологических солевых растворах;
б) адсорбция на стекло или пластик либо неспецифическое связывание с исследуемой матрицей может снижать концентрацию исследуемого вещества в инкубационном или перфузионном растворе;
в) концентрации исследуемого вещества могут быть ограничены его цитотоксическими или физико-химическими свойствами, нарушающими целостность клеточной мембраны, что не позволит достичь электрофизиологических конечных точек;
г) клетки и ткани сердца обладают низкими метаболическими возможностями для метаболизма лекарственных препаратов, поэтому исследования in vitro с использованием исходного вещества не позволяют получить данные о влиянии метаболитов. Если доклинические или клинические исследования in vivo обнаруживают удлинение интервала QT, не согласующееся с результатами исследований in vitro с использованием исходного вещества, следует предусмотреть проведение исследований метаболитов в тест-системах in vitro.
33. При разработке новых технологий анализа калиевых каналов, которые могут быть использованы для предварительного скрининга исследуемых веществ в целях выявления "соединений - ведущих кандидатов", следует подтвердить согласованность новых и стандартных технологий перед началом использования таких новых технологий в целях экспертизы и регистрации лекарственного препарата.
34. Допускается использование протоколов конкурентного связывания, в которых исследуемые вещества изучаются на предмет их способности вытеснять радиоактивно меченый блокатор hERG-канала в клеточной линии, экспрессирующей hERG. Вместе с тем конкуренция за участки связывания с радиолигандами не позволяет получить сведения об агонистических или антагонистических эффектах испытуемого вещества на IKr. Более того, этот анализ не будет выявлять исследуемые вещества, связывающиеся с hERG в участках, отличных от участков связывания радиолиганда. Принимая во внимание потенциальное ограничение, такой анализ не следует считать заменой анализам фиксации потенциала (напряжения), описанным в пунктах 29 - 33 настоящих Указаний.
Электрофизиологические исследования in vivo
35. Модели in vivo с интактными животными позволяют изучить реполяризацию желудочков или ассоциированные с ней аритмии, а также оценить интегральное влияние всего комплекса ионных каналов и типов клеток. Кроме того, интактные животные позволяют изучить потенциальный нейрональный и гормональный ответ на фармакодинамический эффект лекарств.
36. Длина интервала QT на электрокардиограмме - наиболее часто используемая конечная точка, измеряющая влияние исследуемого вещества на реполяризацию желудочков. В специализированных электрофизиологических исследованиях сведения о реполяризации желудочков (например, продолжительность монофазного потенциала действия и эффективный рефрактерный период) можно также получить на моделях in vivo. Одновременно можно оценить дополнительные представляющие интерес параметры безопасности, включая артериальное давление, ритм сердца, интервал PR, ширину комплекса QRS и аритмии.
37. Интервал QT и ритм сердца имеют обратную (нелинейную) зависимость, варьируются между видами и между особями одного вида. Таким образом, изменение ритма сердца само по себе влияет на интервал QT, что может искажать оценку влияния исследуемого вещества на реполяризацию желудочков сердца и интервал QT. Кроме того, следует учитывать вариабельность ритма сердца между особями: во-первых, в силу разницы в автономном тонусе и, во-вторых, вследствие влияния исследуемого вещества на ритм сердца. В связи с этим при интерпретации данных, полученных от тест-систем in vivo, необходимо учитывать одновременные изменения ритма сердца. В идеале данные об интервале QT, получаемые после введения исследуемого вещества, следует сравнивать с контрольными и исходными данными при сопоставимых ритмах сердца. Если вариабельность ритма сердца не обусловлена исследуемым веществом, ее можно снизить путем акклиматизации или при использовании животных моделей, в отношении которых выполнена анестезия. Если эффекты обусловлены исследуемым веществом, наиболее распространенным подходом является коррекция интервала QT на ритм сердца (QTc) с использованием формулы Базетта, Фредерика и других подходов. Выбор формулы коррекции ритма сердца следует обосновать данными, полученными от тест-системы. Если различия в ритме сердца между вмешательством и контролем велики, то корректирующие формулы могут оказаться неэффективными для оценки риска удлинения интервала QT. Альтернативным подходом является поддержание постоянного ритма сердца с помощью искусственного водителя ритма. Иногда целесообразнее провести анализ отношения 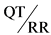 , включая коррекцию интервала QT с использованием формул для отдельных экспериментальных животных.
, включая коррекцию интервала QT с использованием формул для отдельных экспериментальных животных.
38. К лабораторным видам животных, используемым в электрофизиологических исследованиях in vivo, относятся собаки, обезьяны, свиньи, кролики, хорьки и морские свинки. Ионные механизмы реполяризации у взрослых крыс и мышей отличаются от ионных механизмов реполяризации более крупных видов животных, а также человека (основные ионные токи, контролирующие реполяризацию у взрослых крыс и мышей, - Ito), поэтому использование крыс и мышей неприемлемо. Следует выбрать наиболее подходящие тест-системы in vivo и обосновать их выбор.
39. Диапазон доз должен согласовываться с диапазоном, который был использован в основной батарее исследований фармакологической безопасности, и во всех выполнимых случаях включать или превышать ожидаемую экспозицию у человека. Диапазон доз может ограничиваться непереносимостью исследуемого вещества животными (например, рвотой, тремором или гиперактивностью). При проведении исследований, направленных на соотнесение степени замедления реполяризации желудочков с концентрациями исходного исследуемого вещества и его метаболитов, можно использовать контролируемую экспозицию с помощью постоянной внутривенной инфузии. Мониторинг экспозиции исследуемого вещества и метаболитов позволяет интерпретировать данные о зависимости "доза - ответ" и "концентрация - ответ" и планировать последующие исследования (если целесообразно).
40. При проведении исследований и интерпретации результатов учитываются следующие факторы:
а) метод сбора и анализа данных;
б) чувствительность и воспроизводимость тест-систем;
в) период дозирования и измеряемые точки;
г) ритм сердца и другие эффекты, искажающие интерпретацию данных об интервале QT;
д) межвидовые и половые различия (например, электрофизиология сердца, гемодинамика или метаболизм исследуемых веществ);
е) способность исследуемых веществ, влияющих на несколько ионных каналов, давать сложный вид зависимости "доза - ответ" в эксперименте, который может плохо поддаваться интерпретации.
Имитация патологических состояний и аритмий
41. Точная зависимость между замедлением реполяризации желудочков сердца, индуцируемым исследуемым веществом и риском аритмии неизвестна. Целесообразно напрямую изучить аритмогенный риск лекарственных препаратов, удлиняющих интервал QT. При оценке аритмогенного действия могут использоваться животные модели и показатели аритмогенной активности (например, электрическая нестабильность, временная и (или) пространственная дисперсия рефрактерности, обратная частотная зависимость, изменения конфигурации потенциала действия). Приветствуются разработка заинтересованными сторонами описанных моделей и оценка их пригодности при определении риска для человека.