"Методические рекомендации по подготовке разработчиками и производителями лекарственных препаратов, находящихся в обращении на территории Российской Федерации, периодических отчетов по безопасности лекарственных препаратов"
Утверждаю
Врио руководителя
Федеральной службы по надзору
в сфере здравоохранения
М.А.МУРАШКО
04.06.2013
МЕТОДИЧЕСКИЕ РЕКОМЕНДАЦИИ
ПО ПОДГОТОВКЕ РАЗРАБОТЧИКАМИ И ПРОИЗВОДИТЕЛЯМИ
ЛЕКАРСТВЕННЫХ ПРЕПАРАТОВ, НАХОДЯЩИХСЯ В ОБРАЩЕНИИ
НА ТЕРРИТОРИИ РОССИЙСКОЙ ФЕДЕРАЦИИ, ПЕРИОДИЧЕСКИХ
ОТЧЕТОВ ПО БЕЗОПАСНОСТИ ЛЕКАРСТВЕННЫХ ПРЕПАРАТОВ
УВЕДОМЛЕНИЕ
Настоящие методические рекомендации не являются нормативным правовым актом.
Данный документ основывается на подходах Федеральной службы по надзору в сфере здравоохранения к рассматриваемой проблеме и содержит разъяснения по практическим вопросам соблюдения требований законодательства Российской Федерации в сфере осуществления мониторинга безопасности лекарственных средств.
Отклонение от положений методических рекомендаций при условии соблюдения законодательства Российской Федерации не влечет за собой наступления административной или иной ответственности.
Использованные сокращения
АИС Росздравнадзора - Автоматизированная информационная система Росздравнадзора
МНН - международное непатентованное название
Н/П - несерьезные предвиденные [нежелательные реакции]
Н/НП - несерьезные непредвиденные [нежелательные реакции]
ПОБЛП - периодический отчет по безопасности лекарственного препарата
С/П - серьезные предвиденные [нежелательные реакции]
С/НП - серьезные непредвиденные[нежелательные реакции]
DDD - Установленная суточная доза лекарственного препарата (Defined Daily Dose)
Введение
В соответствии с Федеральным законом от 12.04.2010 N 61-ФЗ "Об обращении лекарственных средств" лекарственные препараты, находящиеся в обращении на территории Российской Федерации, подлежат мониторингу безопасности в целях выявления возможных негативных последствий их применения, предупреждения и защиты пациентов от их использования.
Государственная функция по мониторингу безопасности лекарственных препаратов осуществляется путем сбора, обработки и анализа сведений по безопасности лекарственных средств в целях выявления информации о побочных действиях, не указанных в инструкции по применению лекарственного препарата, серьезных нежелательных реакциях, непредвиденных нежелательных реакциях, особенностях его взаимодействия с другими лекарственными препаратами (далее - нежелательные реакции), которые могут представлять угрозу жизни или здоровью пациентов.
В рамках реализации данной государственной функции приказом Министерства здравоохранения и социального развития Российской Федерации от 26.08.2010 N 757н "Об утверждении порядка осуществления мониторинга безопасности лекарственных препаратов для медицинского применения, регистрации побочных действий, серьезных нежелательных реакций, непредвиденных нежелательных реакций при применении лекарственных препаратов для медицинского применения" (зарегистрирован Министерством юстиции Российской Федерации 31.08.2010 N 18324) в отношении разработчиков и производителей лекарственных препаратов, на имя которых выдано регистрационное удостоверение лекарственного препарата (далее - держателей регистрационного удостоверения) установлена обязанность в установленные сроки представлять в Федеральную службу по надзору в сфере здравоохранения (далее - Росздравнадзор) информацию обо всех случаях побочных действий, не указанных в инструкции по применению лекарственного препарата, о серьезных нежелательных реакциях, непредвиденных нежелательных реакциях при применении лекарственных препаратов, об особенностях взаимодействия лекарственных препаратов с другими лекарственными препаратами, а также периодические отчеты по безопасности лекарственных препаратов (далее - ПОБЛП).
В настоящих методических рекомендациях рассмотрены вопросы подготовки и представления ПОБЛП, включая вопросы определения сроков их представления, объема информации по безопасности лекарственных препаратов, подлежащей включению в ПОБЛП, структуре и формату ПОБЛП, а также порядка направления отчетов в Росздравнадзор.
Рекомендации разработаны на основе:
1. Федерального закона от 12.04.2010 N 61-ФЗ "Об обращении лекарственных средств";
2. Федерального закона от 21.11.2011 N 323-ФЗ "Об основах охраны здоровья граждан";
3. Федерального закона от 27.07.2006 N 152-ФЗ "О персональных данных";
4. Постановления Правительства Российской Федерации от 30.06.2004 N 323 "Об утверждении положения о Федеральной службе по надзору в сфере здравоохранения";
5. Приказа Министерства здравоохранения и социального развития Российской Федерации от 26.08.2010 N 757н "Об утверждении порядка осуществления мониторинга безопасности лекарственных препаратов для медицинского применения, регистрации побочных действий, серьезных нежелательных реакций, непредвиденных нежелательных реакций при применении лекарственных препаратов для медицинского применения" (зарегистрирован Министерством юстиции Российской Федерации 31.08.2010 N 18324);
6. Приказа Министерства здравоохранения и социального развития Российской Федерации от 26.08.2010 N 758н "Об утверждении Порядка приостановления применения лекарственного препарата для медицинского применения" (зарегистрирован Министерством юстиции Российской Федерации 31.08.2010 N 18325).
В процессе разработки настоящих методических рекомендаций приняты во внимание положения следующих документов:
- Гармонизированные Трехсторонние Руководства Международной конференции по гармонизации (ICH)
- Первого и второго пересмотра рекомендаций ICH E2C: (R1) "Управление данными по клинической безопасности: Периодические отчеты по безопасности лекарственных средств, находящихся на рынке" <1> и (R2) Международной конференции по гармонизации ICH E2C (R2) "Периодический отчет о соотношении пользы и риска лекарственного препарата" <2>
--------------------------------
<1> ICH Harmonized Tripartite Guideline E2C (R1) "Clinical safety data management: periodic safety update reports for marketed drugs".
<2> ICH Harmonized Tripartite Guideline E2C (R2)"Periodic Risk-Benefit Evaluation Report (PBER)".
- ICH E2D "Управление данными по безопасности в пострегистрационном периоде: определения и стандарты срочной отчетности" <1>,
--------------------------------
<1> ICH Harmonized Tripartite Guideline E2D "Post-approval safety data management: definitions an standards for expedited reporting" E.
- ICH E2E "Планирование мероприятий по фармаконадзору" <1>
--------------------------------
<1> ICH Harmonized Tripartite Guideline ICH E2E "Pharmacovigilance planning".
- а также подготовленные Европейской Комиссией Европейского Союза
- Тома 9A Правил обращения лекарственных препаратов в Европейском Союзе "Рекомендации по мониторингу безопасности лекарственных средств, предназначенных для человека" <1>;
--------------------------------
<1>" The Rules Governing Medicinal Products in the European Union, Volume 9a, "Guidelines on Pharmacovigilance for Medicinal Products for Human Use".
- Руководств Европейского Агентства по лекарственным средствам по надлежащей практике фармаконадзора <1>.
--------------------------------
<1> EMA Good Pharmacovigilance Practice Modules.
В настоящих методических рекомендациях изложены базовые принципы подготовки ПОБЛП. По дополнительным вопросам и разъяснениям, связанным с подготовкой ПОБЛП, рекомендуется обращаться в Федеральную службу в сфере здравоохранения.
Термины и определения
1. Лекарственные средства - вещества или их комбинации, вступающие в контакт с организмом человека или животного, проникающие в органы, ткани организма человека или животного, применяемые для профилактики, диагностики (за исключением веществ или их комбинаций, не контактирующих с организмом человека или животного), лечения заболевания, реабилитации, для сохранения, предотвращения или прерывания беременности и полученные из крови, плазмы крови, из органов, тканей организма человека или животного, растений, минералов методами синтеза или с применением биологических технологий. К лекарственным средствам относятся фармацевтические субстанции и лекарственные препараты.
2. Лекарственные препараты - лекарственные средства в виде лекарственных форм, применяемые для профилактики, диагностики, лечения заболевания, реабилитации, для сохранения, предотвращения или прерывания беременности.
3. Оригинальное лекарственное средство - лекарственное средство, содержащее впервые полученную фармацевтическую субстанцию или новую комбинацию фармацевтических субстанций, эффективность и безопасность которых подтверждены результатами доклинических исследований лекарственных средств и клинических исследований лекарственных препаратов.
4. Воспроизведенное лекарственное средство - лекарственное средство, содержащее такую же фармацевтическую субстанцию или комбинацию таких же фармацевтических субстанций в такой же лекарственной форме, что и оригинальное лекарственное средство, и поступившее в обращение после поступления в обращение оригинального лекарственного средства.
5. Международное непатентованное наименование (далее - МНН) лекарственного средства - наименование фармацевтической субстанции, рекомендованное Всемирной организацией здравоохранения.
6. Торговое наименование лекарственного средства - наименование лекарственного средства, присвоенное его разработчиком. Важно обратить внимание, что в практике подготовки ПОБЛП под торговым наименованием подразумевается наименование лекарственного препарата, указанное в российском регистрационном удостоверении.
7. Качество лекарственного средства - соответствие лекарственного средства требованиям фармакопейной статьи либо в случае ее отсутствия нормативной документации или нормативного документа.
8. Безопасность лекарственного средства - характеристика лекарственного средства, основанная на сравнительном анализе его эффективности и риска причинения вреда здоровью.
9. Эффективность лекарственного препарата - характеристика степени положительного влияния лекарственного препарата на течение, продолжительность заболевания или его предотвращение, реабилитацию, на сохранение, предотвращение или прерывание беременности.
10. Субъекты обращения лекарственных средств - физические лица, в том числе индивидуальные предприниматели, и юридические лица, осуществляющие деятельность при обращении лекарственных средств.
11. Разработчик лекарственного средства - организация, обладающая правами на результаты доклинических исследований лекарственного средства, клинических исследований лекарственного препарата, а также на технологию производства лекарственного средства.
12. Производитель лекарственных средств - организация, осуществляющая производство лекарственных средств в соответствии с требованиями Федерального закона от 12.04.2010 N 61-ФЗ "Об обращении лекарственных средств".
13. Доклиническое исследование лекарственного средства биологические, микробиологические, иммунологические, токсикологические, фармакологические, физические, химические и другие исследования лекарственного средства путем применения научных методов оценок в целях получения доказательств безопасности, качества и эффективности лекарственного средства.
14. Клиническое исследование лекарственного препарата - изучение диагностических, лечебных, профилактических, фармакологических свойств лекарственного препарата в процессе его применения у человека, животного, в том числе процессов всасывания, распределения, изменения и выведения, путем применения научных методов оценок в целях получения доказательств безопасности, качества и эффективности лекарственного препарата, данных о нежелательных реакциях организма человека, животного на применение лекарственного препарата и об эффекте его взаимодействия с другими лекарственными препаратами и (или) пищевыми продуктами, кормами.
15. Пострегистрационное клиническое исследование лекарственного препарата для медицинского применения - клиническое исследование лекарственного препарата для медицинского применения, проводимое производителем лекарственного препарата, гражданский оборот которого осуществляется после государственной регистрации, в целях дополнительного сбора данных о его безопасности и эффективности, расширения показаний к применению данного лекарственного препарата, а также выявления нежелательных реакций пациентов на его действие.
16. Побочное действие - реакция организма, возникшая в связи с применением лекарственного препарата в дозах, рекомендуемых в инструкции по его применению, для профилактики, диагностики, лечения заболевания или для реабилитации.
17. Серьезная нежелательная реакция - нежелательная реакция организма, связанная с применением лекарственного препарата, приведшая к смерти, врожденным аномалиям или порокам развития либо представляющая собой угрозу жизни, требующая госпитализации или приведшая к стойкой утрате трудоспособности и (или) инвалидности.
18. Непредвиденная нежелательная реакция - нежелательная реакция организма (в том числе связанная с применением лекарственного препарата в соответствии с инструкцией по его применению), сущность и тяжесть которой не соответствуют информации о лекарственном препарате, содержащейся в инструкции по его применению.
1. Общие вопросы подготовки ПОБЛП
Получение достаточной информации о профиле эффективности и безопасности лекарственного препарата невозможно без выявления и анализа данных о его нежелательных реакциях и особенностях взаимодействия с другими лекарственными препаратами, пищевыми продуктами, а также биологически активными добавками к пище в пострегистрационном периоде. ПОБЛП является аналитическим обзором сведений о безопасности лекарственного препарата, позволяющим уточнить оценку соотношения пользы и риска применения лекарственного препарата в медицинской практике.
Учитывая это, ПОБЛП могут предоставляться в Росздравнадзор в трех основных форматах, обеспечивающих достаточный охват и уровень анализа информации о безопасности лекарственных препаратов, полученной в пострегистрационном периоде:
- Формат, описанный в руководстве Международной конференции по гармонизации ICH E2C (R1) "Управление данными по клинической безопасности: Периодические отчеты по безопасности лекарственных средств, находящихся на рынке" <1>
--------------------------------
<1> ICH Harmonized Tripartite Guideline E2C (R1) "Clinical safety data management: periodic safety update reports for marketed drugs".
Формат, описанный в руководстве Международной конференции по гармонизации ICH E2C (R2) "Периодический отчет о соотношении пользы и риска лекарственного препарата" <1>
--------------------------------
<1> ICH Harmonized Tripartite Guideline E2C (R2)"Periodic Risk-Benefit Evaluation Report (PBER)".
Формат ПОБЛП, описанный в разделах 1.1 - 3 настоящих рекомендаций.
Предлагаемый в настоящих рекомендациях формат ПОБЛП является адаптированной версией формата ПОБЛП, описанного в Руководстве Международной конференции по гармонизации (ICH) E2C (R1) "Управление данными по клинической безопасности: Периодические отчеты по безопасности лекарственных средств, находящихся на рынке".
1.1 Объем информации по безопасности лекарственного препарата, рекомендуемый к включению в ПОБЛП
В пострегистрационном периоде держатель регистрационного удостоверения получает данные по безопасности лекарственного препарата из следующих источников:
1. Сообщения субъектов обращения лекарственных средств о выявленных нежелательных реакциях (спонтанные сообщения), включая:
a. Сообщения специалистов здравоохранения (медицинских работников);
b. Сообщения о нежелательных реакциях, поступающие от органов управления здравоохранением субъектов Российской Федерации;
c. Сообщения лиц, не являющихся специалистами здравоохранения, включая пациентов или потребителей;
d. Сообщения юридических лиц, осуществляющих обращение данного лекарственного препарата за рубежом, в рамках контрактных соглашений.
2. Новые данные по безопасности и эффективности, полученные в ходе доклинических, клинических, эпидемиологических исследований, спонсором которых является держатель регистрационного удостоверения лекарственного препарата.
В эту группу могут быть включены данные регистров заболевания или использования лекарственного препарата (например, орфанного лекарственного средства).
3. Новые данные по безопасности и эффективности из научной литературы.
4. Сообщения о решениях зарубежных регуляторных органов об изменении регистрационного статуса лекарственного препарата, изменениях регистрационной документации, связанные с безопасностью, либо иных мерах, направленных на обеспечение безопасности его использования (например, программы управления рисками).
5. Другие источники информации, включая сведения, полученные от медицинских организаций, региональных центров мониторинга безопасности лекарственных препаратов, правоохранительных органов о фактах вреда жизни и здоровью человека вследствие применения лекарственного препарата (включая случаи намеренной или непреднамеренной передозировки, злоупотребления или использования в противоправных целях).
Данные сведения составляют основу периодического отчета по безопасности.
В ПОБЛП следует включать информацию обо всех нежелательных реакциях, вне зависимости от того, считает ли держатель регистрационного удостоверения эти реакции связанными с применением данного лекарственного препарата.
Порядок оценки причинно-следственной связи между применением лекарственного препарата и развитием нежелательной реакции описан в Методических рекомендациях Росздравнадзора от 02.10.2008 "Определение степени достоверности причинно-следственной связи "Неблагоприятная побочная реакция - лекарственное средство" (классификация и методы)". <1>
--------------------------------
<1> Опубликованы на сайте Росздравнадзора (раздел "Лекарственные средства", подраздел "Мониторинг безопасности лекарственных средств, находящихся в обращении на территории Российской Федерации", рубрика "Методические разработки").
Держатель регистрационного удостоверения может принять решение не включать в ПОБЛП те нежелательные реакции, причинно-следственная связь которых с применением препарата отрицается одновременно отправителем спонтанного сообщения и держателем регистрационного удостоверения.
В целях обеспечения необходимой полноты информации о безопасности лекарственного препарата, полученной в пострегистрационном периоде, в ПОБЛП следует включать данные, полученные от иных юридических лиц, осуществляющих обращение данного лекарственного препарата за пределами Российской Федерации в рамках контрактных обязательств (включая сведения из спонтанных сообщений, клинических исследований и др.).
Указанные выше сведения по безопасности лекарственного препарата должны быть проанализированы держателем регистрационного удостоверения в целях оценки их влияния на соотношение пользы и риска использования лекарственного средства, а также необходимости принятия дополнительных мер, направленных на повышение безопасности его применения (изменение инструкции, рецептурного статуса, распространения информационных писем для специалистов здравоохранения).
В ПОБЛП должны быть приведены данные о предполагаемом объеме потребления препарата (экспозиции), что позволит количественно оценить риски развития нежелательных реакций или проблем безопасности (см. раздел 3.5).
1.2 Предоставление данных об отсутствии или недостаточности ожидаемого терапевтического эффекта лекарственных препаратов, применяемых в терапии жизнеугрожающих заболеваний
С учетом клинической значимости отсутствие или недостаточная клиническая эффективность лекарственных препаратов, применяемых для лечения жизнеугрожающих заболеваний, может попадать под определение серьезной нежелательной реакции, приведенное в Федеральном законе от 12.04.2010 N 61-ФЗ "Об обращении лекарственных средств".
В связи с этим информацию о случаях отсутствия ожидаемого терапевтического эффекта рекомендуется включать в особый раздел ПОБЛП (см. Раздел 3.10).
1.3 Временной охват информации
В ПОБЛП включается клиническая и доклиническая информация о безопасности лекарственного препарата, которая получена держателем регистрационного удостоверения в период сбора сведений для ПОБЛП. Исключения составляют следующие данные:
- В каждый отчет включается вся информация об изменениях регистрационного статуса лекарственного препарата в Российской Федерации и за рубежом, а также изменениях регистрационной документации, связанных с его безопасностью, которые произошли за весь пострегистрационный период, начиная со дня первой государственной регистрации данного лекарственного препарата в мире;
- В каждый ПОБЛП включаются обобщенные сведения обо всех выявленных за весь пострегистрационный период нежелательных реакциях, которые являются одновременно серьезными и непредвиденными (в форме обзора эпизодов выявления данных реакций, а также последовательное описание действий, предпринятых держателем регистрационного удостоверения / регуляторными агентствами в связи с обнаружением этих реакций (например, проведение дополнительных исследований, внесения изменений в инструкцию, приостановления применения лекарственного препарата).
1.4 Единый периодический отчет по безопасности для лекарственных препаратов, содержащих одно активное вещество
Держателям регистрационных удостоверений лекарственных препаратов рекомендуется готовить один периодический отчет на лекарственные препараты, содержащие одно и то же действующее вещество (или комбинацию действующих веществ) в разных лекарственных формах, дозировках и составах.
Данные по безопасности комбинированных лекарственных препаратов, действующие вещества которых также используются в монопрепаратах, выпускаемых держателем регистрационного удостоверения, могут быть представлены в отдельном ПОБЛП или в особом разделе периодического отчета по безопасности препарата, содержащего одно из действующих веществ данной комбинации. Вопрос о структуре ПОБЛП в этом случае следует решать, исходя из значимости различий фармакологических свойств препарата, включая различия показаний к применению и профиля безопасности монопрепарата и комбинированного лекарственного препарата. Если данные по безопасности комбинированного препарата и монопрепарата представляются в двух независимых ПОБЛП, данные отчеты должны быть снабжены перекрестными ссылками, облегчающими поиск значимой информации по безопасности действующих веществ.
В случае если показания к применению разных лекарственных форм значительно различаются <1>, может оказаться целесообразным представить данные по безопасности соответствующих лекарственных препаратов в самостоятельных разделах одного ПОБЛП.
--------------------------------
<1> Например, если один и тот же лекарственный препарат в форме шипучих таблеток показан к применению у взрослых, а в форме сиропа - у детей.
1.5 Представление отчета вне зависимости от фактического обращения лекарственного препарата на фармацевтическом рынке Российской Федерации
Новая информация об эффективности и безопасности лекарственных препаратов появляется непрерывно.
Сведения о нежелательных реакциях, а также иных факторах риска применения лекарственных средств содержатся не только в спонтанных сообщениях, поступающих держателю регистрационного удостоверения, но и в материалах доклинических, клинических исследований, данных ретроспективного изучения его использования, научных статьях. Учитывая это, ПОБЛП следует представлять в установленные сроки (см. ниже) вне зависимости от фактического обращения зарегистрированного лекарственного препарата на фармацевтическом рынке Российской Федерации.
1.6 Принципы оценки предвиденности нежелательных реакций
Для оценки предвиденности нежелательной реакции рекомендуется использовать ту редакцию инструкции, которая была утверждена в Российской Федерации на дату начала отчетного периода сбора данных для ПОБЛП.
Соответственно реакции, сведения о которых были внесены в инструкцию по применению только в период сбора данных для данного ПОБЛП, в этом отчете будут описываться как непредвиденные.
Информацию о возможном изменении частоты нежелательных реакций по сравнению с описанной в инструкции по медицинскому применению рекомендуется классифицировать как непредвиденную нежелательную реакцию. Также в ПОБЛП следует дать разъяснения держателя регистрационного удостоверения относительно причин изменения частоты нежелательных реакций (изменение целевой популяции, у которой применялся препарат, превышение дозировки, проблемы качества лекарственного препарата и др.).
В случае, если для оценки предвиденности реакций держатель регистрационного удостоверения руководствуется Базовой характеристикой лекарственного препарата (Company Core Safety Datasheet, CCSDS) или основными сведениями по безопасности лекарственного препарата (Company Core Safety Infromation, CCSI), то копии указанных документов прикладываются к ПОБЛП.
1.7 Использование литературных источников
Держателям регистрационных удостоверений рекомендуется создать перечень литературных источников (отечественных и зарубежных рецензируемых научных журналов, доступных в сети Интернет или на бумажном носителе), которые будут регулярно мониторироваться в целях оценки безопасности выпускаемых лекарственных препаратов.
Выбор литературных источников для подобного мониторинга должен производиться с учетом сферы применения лекарственного средства.
Перечень мониторируемых научных литературных источников рекомендуется прикладывать к ПОБЛП.
1.8 Получение информации о решениях зарубежных регуляторных органов, принятых в связи с проблемами безопасности лекарственных препаратов
Наряду с литературными данными рекомендуется отслеживать решения зарубежных регуляторных агентств по ограничению или изменению порядка обращения лекарственных препаратов. Возможные источники информации по регуляторным решениям в области безопасности лекарственных средств указаны в приложении 1.
1.9 Описание нежелательных реакций на лекарственный препарат
Для описания нежелательных реакций рекомендуется использовать единообразную терминологию. Для этой цели возможно использовать термины справочников информационного ресурса по фармаконадзору Автоматизированной информационной системы Росздравнадзора (далее - АИС Росздравнадзора), которые являются русскими эквивалентами Терминологии нежелательных реакций, разработанной Всемирной Организацией Здравоохранения (WHO-ART). Также держатели регистрационных удостоверений при описании нежелательных реакций могут руководствоваться самостоятельно переведенными терминами словаря регуляторной медицинской терминологии ICH (MedDRA).
Показания к применению лекарственного препарата рекомендуется указывать в соответствии с Международной классификацией болезней Всемирной Организации Здравоохранения X-го пересмотра, введенной в учреждениях здравоохранения Российской Федерации приказом Минздрава России от 27.05.1997 N 170 "О переходе органов и учреждений здравоохранения Российской Федерации на международную статистическую классификацию болезней и проблем, связанных со здоровьем, X пересмотра".
В случае если в сообщении отсутствует диагноз заболевания, для терапии которого назначался лекарственный препарат, держатель регистрационного удостоверения может самостоятельно указать предполагаемый диагноз, исходя из анализа доступных данных. Такое предположение держателя регистрационного удостоверения должно сопровождаться соответствующим примечанием.
В случае невозможности точно описать или классифицировать описываемую нежелательную реакцию допустимо использование терминологии отправителя, указав ее в кавычках.
При разногласиях держателя регистрационного удостоверения и отправителя сообщения относительно описания, исхода, причины нежелательной реакции, показания к назначению лекарственного препарата позиция держателя регистрационного удостоверения по данному вопросу может быть приведена в виде особого примечания или комментария в том разделе ПОБЛП, где описана соответствующая реакция.
2. Определение даты представления ПОБЛП
В соответствии с приказом Минздравсоцразвития России от 26.08.2010 N 757н периодические отчеты направляются держателем регистрационного удостоверения лекарственного препарата, на имя которого выдано регистрационное удостоверение лекарственного препарата, в Федеральную службу по надзору в сфере здравоохранения на электронном и бумажном носителе в сроки, отсчитываемые от даты регистрации лекарственного препарата в стране, где лекарственный препарат был впервые разрешен к медицинскому применению:
- в течение первых двух лет регистрации лекарственного препарата - каждые 6 месяцев;
- в течение последующих двух лет - третьего и четвертого года регистрации лекарственного препарата - ежегодно;
- начиная с пятого года регистрации лекарственного препарата - один раз в три года.
Периодические отчеты предоставляются не позднее 30 дней от даты окончания срока отсчета.
Порядок определения даты первой регистрации лекарственного препарата приведен в схеме.
1. Сроки предоставления ПОБЛП воспроизведенных лекарственных препаратов отсчитываются от даты первой регистрации оригинального лекарственного препарата с активным веществом (комбинацией активных веществ) данного международного непатентованного или группировочного названия (за исключением биологических лекарственных препаратов).
2. Сроки предоставления ПОБЛП для биологических лекарственных средств (иммунобиологических лекарственных средств, лекарственных средств, полученных из крови и плазмы крови человека, лекарственных средств, произведенных путем биотехнологических процессов (технологий и методов), генотерапевтических и соматотерапевтических лекарственных средств, содержащих действующие вещества биологического происхождения), а также биоаналоговых лекарственных средств (лекарственных средств, схожих с оригинальным биологическим лекарственным средством по технологии производства, фармацевтической субстанции (комбинации фармацевтических субстанций), лекарственной форме, показаниям к применению и поступившее в обращение с соблюдением интеллектуальных прав на оригинальное лекарственное средство) отсчитываются от даты регистрации данного лекарственного препарата в Российской Федерации.
3. Дату отсчета сроков представления ПОБЛП возможно округлить до первого числа месяца, следующего за датой первичной регистрации оригинального лекарственного препарата.
4. Для лекарственного препарата, выпускаемого в нескольких лекарственных формах, срок представления ПОБЛП отсчитывается от даты регистрации первой лекарственной формы оригинального лекарственного препарата в порядке, приведенном в пунктах 5 - 10.
5. В случае если оригинальный лекарственный препарат с активным веществом (комбинацией активных веществ) данного международного непатентованного или группировочного названия (далее - оригинальный лекарственный препарат) впервые в медицинской практике был зарегистрирован (разрешен к применению в медицинской практике) в СССР до 01.01.1992, в качестве даты отсчета срока предоставления ПОБЛП оригинального лекарственного препарата и воспроизведенных препаратов используется дата приказа о первой регистрации оригинального препарата (дата выдачи первого регистрационного удостоверения). Сведения о приказах Минздрава СССР о разрешении к медицинскому применению лекарственных препаратов содержатся, в частности, в справочных правовых информационных системах ("Гарант", "Консультант" и др.).
6. В случае если оригинальный лекарственный препарат был зарегистрирован в государствах Европейского Союза ранее, чем в Российской Федерации, в качестве даты, от которой отсчитываются сроки представления ПОБЛП, рекомендуется использовать так называемую "европейскую дату рождения препарата" (European Birthdate), опубликованную на сайте Европейского Медицинского Агентства (ЕМА). <1>
--------------------------------
<1> http://www.ema.europa.eu/ema/doc_index.jsp?curl=pages/includes/document/document_detail.jsp?webContentId=WC500124999&murl=menus/document_library/document_library.jsp&mid=0b01ac058009a3dc
7. В случае если лекарственный препарат был впервые зарегистрирован за пределами Российской Федерации, но не в государствах ЕС, для определения даты отсчета сроков представления ПОБЛП предпочтительно использовать дату первичной регистрации в США. Данная информация доступна на сайте Управления США по контролю за продуктами питания и лекарственными средствами (Food and Drug Administration) <1>.
--------------------------------
<1> Раздел "информация FDA о лекарственных средствах" "Drugs@FDA" по адресу раздел Drugs@FDA, http://www.accessdata.fda.gov/scripts/cder/drugsatfda/).
8. В случае если оригинальный лекарственный препарат впервые зарегистрирован за пределами Российской Федерации, но не в странах ЕС или США, для определения даты первичной регистрации возможно использовать информацию сайтов ведущих мировых регуляторных агентств, включая Агентство по здравоохранению Канады (Health Canada) (http://webprod3.hc-sc.gc.ca/dpd-bdpp/index-eng.jsp), Управление Австралии по медицинской продукции (Therapeutic Goods Administration Australia https://www.ebs.tga.gov.au/) и других регуляторных агентств.
9. При отсутствии информации о дате первой регистрации на информационных ресурсах зарубежных регуляторных органов в сфере обращения лекарственных препаратов, дата первичной регистрации оригинального препарата может быть установлена на основе данных научной литературы. В этом случае в разделе ПОБЛП, посвященном международному регуляторному статусу препарата, приводится ссылка на источник литературных данных о первичной регистрации препарата.
10. В случае если информация о регистрации лекарственного препарата отсутствует на перечисленных информационных ресурсах зарубежных регуляторных органов, а также в научной литературе (например, для препаратов, традиционно применяемых в медицинской практике или используемых более 50 лет), держатель регистрационного удостоверения может разработать собственный график предоставления ПОБЛП по данному лекарственному средству. Данный график рекомендуется предварительно согласовать с Федеральной службой по надзору в сфере здравоохранения.
Схема. Рекомендуемый порядок определения даты отсчета сроков представления ПОБЛП:
3. Рекомендуемый формат периодического отчета по безопасности лекарственного препарата
Рекомендуется использовать следующую структуру ПОБЛП:
- Титульный лист
- Резюме
- Оглавление
- Введение
- Сведения о регистрации лекарственного препарата в Российской Федерации и других странах
- Сведения об изменении регистрационного статуса и регистрационной документации в связи с проблемами безопасности
- Данные об объеме потребления лекарственного препарата
- В клинических исследованиях
- В медицинской практике
- Информация о выявленных нежелательных реакциях и проблемах безопасности
- Общие данные о выявленных нежелательных реакциях
- Анализ отдельных нежелательных реакций, имеющих особую клиническую значимость
- летальные нежелательные реакции
- серьезные непредвиденные реакции (комплексный анализ в сочетании с данными предыдущих ПОБЛП)
- нежелательные реакции, требующие внимания для оценки соотношения пользы и риска
- дополнительная информация по реакциям, описанным в предыдущих ПОБЛП
- Структурированные перечни нежелательных реакций
- Обобщенные таблицы нежелательных реакций
- Информация о безопасности, полученная в ходе клинических исследований/ дальнейшей разработки лекарственного препарата
- Исследования, в которых была получена важная информация по безопасности
- Исследования безопасности лекарственного препарата
- Литературные данные об изучении лекарственного препарата
- Информация, полученная после окончания периода сбора данных для ПОБЛП
- Итоговая оценка безопасности лекарственных препаратов
- Заключение
- Перечень использованной литературы
- Приложения
Рекомендации по подготовке разделов ПОБЛП
3.1. Титульный лист
Рекомендуемый образец оформления титульного листа ПОБЛП
3.2. Резюме периодического отчета
В резюме следует включать следующую информацию:
- Административные данные ПОБЛП (номер, отчетный период, наименование препарата, лекарственные формы, дозировки, реквизиты регистрационных удостоверений)
- Показания к применению (согласно инструкции, действующей на момент завершения подготовки ПОБЛП)
- Количество стран, в которых зарегистрирован лекарственный препарат
- Информация о решениях зарубежных регуляторных агентств по приостановлению, отзыву или ограничению обращения препарата (при наличии)
- Обобщенные сведения об изменениях инструкции по медицинскому применению лекарственного препарата, связанных с безопасностью в Российской Федерации и в других государствах, где лекарственный препарат зарегистрирован, за отчетный период
- Сведения об объемах потребления данного лекарственного препарата в мире за отчетный период (см. глава 3, пункт 3.5.)
- Количество выявленных серьезных и несерьезных нежелательных реакций, информация о которых включена в данный ПОБЛП (общемировые данные)
- Количество нежелательных реакций, поступивших от российских субъектов обращения лекарственных препаратов за отчетный период
- Новые сведения по безопасности, приведенные в ПОБЛП
- Информация об изменении профиля безопасности, а также изменении соотношения пользы и риска
3.3. Сведения о регистрации лекарственного препарата в Российской Федерации и других странах
В этом разделе приводятся сведения о государственной регистрации лекарственного препарата в Российской Федерации и других странах мира с даты первой регистрации.
Информация приводится в хронологическом порядке, определяемом датой регистрации, начиная с даты первого разрешения применения препарата в мировой медицинской практике. Если в одной и той же стране разные лекарственные формы одного препарата зарегистрированы в разное время, то указывают дату первой регистрации.
В разделе "Дата" приводятся (если известны) даты регистрации, продления регистрации, отказов в регистрации, приостановления или аннулирования регистрации, первого фактического поступления препарата на рынок, внесения изменений в регистрационную документацию, связанных с безопасностью. Каждая из дат должна сопровождаться соответствующим пояснением (в скобках).
В разделе "Комментарии" указываются важные отличия инструкции по применению, утвержденной в Российской Федерации, с инструкциями, утвержденными в других странах (рекомендуется указывать различия в разделах "Показания к применению", "Противопоказания", "Способ применения и дозы", "Особые указания", "Побочные действия"). Для удобства изложения и восприятия материала можно использовать несколько таблиц для разных лекарственных форм или дозировок лекарственного препарата (если разные дозировки применяются по различным показаниям).
В случае если лекарственный препарат зарегистрирован в большом количестве стран, таблицу с регистрационными данными рекомендуется включить в ПОБЛП в качестве приложения.
Рекомендуемый формат представления данных о регистрации лекарственного препарата в Российской Федерации и других странах представлен в таблице 1. Пример заполнения данной таблицы приведен в приложении 2.
Таблица 1. Рекомендуемый формат представления данных о регистрации лекарственного препарата в Российской Федерации и других странах.
3.4. Сведения об изменении регистрационного статуса и регистрационной документации в связи с проблемами безопасности
В этот раздел включается подробное описание мер государственных органов исполнительной власти в сфере здравоохранения Российской Федерации, а также зарубежных регуляторных агентств в связи с выявлением проблем безопасности лекарственного препарата.
Раздел охватывает все меры, принятые со дня первой регистрации в мире лекарственного препарата данного держателя регистрационного удостоверения.
Сведения приводятся в хронологическом порядке и не группируются по странам, а приводятся по датам принятия. В скобках, рядом с каждым регуляторным решением, следует указать соответствующую страну, а также дать краткую характеристику причин, приведших к данным регуляторным решениям.
В раздел рекомендуется включать:
- отзывы (аннулирования) или приостановления регистрации (в т.ч. отдельных лекарственных форм или дозировок)
- Изменение порядка отпуска препарата (например, перевод в рецептурный отпуск)
- ограничение применения (противопоказания, в т.ч. для отдельных категорий пациентов, меры предосторожности)
- приостановление клинических исследований.
К ПОБЛП следует приложить копии документов, подтверждающих данные регуляторные решения, копии изменений регистрационной документации, копии писем специалистам здравоохранения по данному вопросу (при наличии), а также другие аналогичные материалы.
3.5. Сведения об объемах потребления лекарственного препарата
В целях обеспечения возможности оценки риска развития нежелательных реакций в пострегистрационном периоде, в ПОБЛП рекомендуется приводить данные об объемах потребления препарата за отчетный период.
Рекомендуется приводить данные об общем объеме потребления лекарственного препарата в мире и отдельно в Российской Федерации.
Также целесообразно привести раздельные данные об объемах потребления препарата в клинических исследованиях (которые можно определить с большой точностью) и в медицинской практике. В случае с слепыми контролируемыми клиническими исследованиями, которые не завершены на момент окончания сбора данных для ПОБЛП, следует привести приблизительные данные об объеме потребления изучаемого лекарственного средства.
Также в данном разделе может быть приведена оценка потребления препарата разными категориями пациентов. Эта информация может быть необходимой в случаях, когда определенная проблема безопасности затрагивает только одну из групп пациентов (например, детей или больных с почечной недостаточностью).
Информация об объемах реализации лекарственного средства в количестве упаковок, массе активного вещества, количестве единиц дозирования не позволяет оценить реальный объем потребления препарата ввиду различия режимов дозировки у разных пациентов, продолжительности курса лечения при разных показаниях и др.
В связи с этим при вычислении объемов потребления лекарственного препарата рекомендуется использовать предложенную ВОЗ методику определения экспозиции лекарственного средства в пациенто-годах на основе установленной суточной дозы лекарственного препарата (Defined Daily Dose, DDD).
Информация о методике подсчета объема потребления лекарственного средства с использованием установленной суточной дозы приведена в приложении 3.
3.6. Общие данные о выявленных нежелательных реакциях
В данном разделе приводится обобщенная информация о нежелательных реакциях, выявленных держателем регистрационного удостоверения лекарственного препарата за отчетный период.
В раздел также можно включить случаи нежелательных реакций на препараты данного МНН, для которых невозможно исключить применение лекарственного препарата держателя регистрационного удостоверения (например, когда отсутствуют сведения о торговом названии, номере серии и т.п.)
Рекомендуется приводить данные об общем количестве выявленных нежелательных реакций в мире. Отдельно (в скобках каждой ячейки таблицы) рекомендуется представлять информацию о количестве нежелательных реакций, выявленных в Российской Федерации.
Важно обратить внимание, что реакции на лекарственные препараты, информация о которых получена из научных публикаций (статей, тезисов, выступлений на научных конференциях) следует включать в ПОБЛП в качестве отдельных эпизодов нежелательных реакций при наличии четырех критериев: идентифицируемый пациент (например, инициалы либо место лечения), симптомы реакции (описанные медицинскими терминами), наименование препарата (торговое наименование или МНН + производитель), наличие автора публикации.
Рекомендуемый формат представления общих сведений о выявленных нежелательных реакциях на лекарственный препарат приведен в таблице 2.
Таблица 2. Рекомендуемый формат представления общих сведений о выявленных нежелательных реакциях на лекарственный препарат.
3.7. Отдельные эпизоды нежелательных реакций, имеющие клиническую значимость
В данном разделе приводится текстовое изложение эпизодов наиболее значимых нежелательных реакций, имеющих большое значение для оценки пользы и риска применения лекарственного препарата.
К таким нежелательным реакциям могут относиться летальные реакции, серьезные и непредвиденные реакции. Кроме того, в разделе может быть приведена дополнительная информация по значимым нежелательным реакциям, описанным в предыдущем ПОБЛП.
При небольшом количестве нежелательных реакций каждая реакция может быть подробно рассмотрена отдельно. В этом случае по структуре и степени детализации сведения об отдельных реакциях могут примерно повторять структуру выписного эпикриза пациента (диагноз, терапия, характеристика реакции, ее течение, исход, данные инструментальных исследований (если применимо).
При большом числе реакций информация о них может быть обобщена в аналитическом обзоре или в виде таблицы.
Необходимо отметить, что в этом разделе могут быть рассмотрены не только реакции, информация о которых получена держателем регистрационного удостоверения от субъектов обращения лекарственных средств, но и данные из научной литературы.
Сведения о нежелательных реакциях, неподтвержденных специалистом здравоохранения, в данном разделе рассматриваются только в случае, если они имеют особую значимость для оценки профиля безопасности лекарственного препарата.
Примерная структура данного раздела, а также пример описания индивидуальной нежелательной реакции приведен в приложениях 4 и 5.
3.8. Структурированные перечни нежелательных реакций и обобщенные таблицы нежелательных реакций
В целях обеспечения возможности всестороннего анализа информации о нежелательных реакциях сведения о них рекомендуется представлять в виде структурированных перечней (см. таблицу 3), позволяющих провести качественную оценку информации, и обобщенных таблиц, необходимых для количественной оценки выявленных рисков.
Структурированные перечни
В форме структурированных перечней (подробное изложение) целесообразно представлять информацию о следующих значимых категориях нежелательных реакций (отдельный перечень для каждой категории):
- Серьезные реакции, информация о которых поступила напрямую держателю регистрационного удостоверения от специалистов здравоохранения
- Несерьезные непредвиденные, информация о которых поступила напрямую держателю регистрационного удостоверения от специалистов здравоохранения
- Серьезные реакции, выявленные в клинических исследованиях, спонсором которых является держатель регистрационного удостоверения
- Серьезные реакции из литературных источников
- Несерьезные непредвиденные реакции из литературных источников
- Серьезные реакции, сведения о которых поступили из органов управления здравоохранением и правоохранительных органов (умышленные передозировки, злоупотребление, использование в противоправных целях).
Таблица 3. Рекомендованный формат структурированного перечня нежелательных реакций.
При заполнении структурированного перечня рекомендуется придерживаться следующих правил:
- Для каждой из указанных выше категорий нежелательных реакций рекомендуется предусмотреть отдельный перечень
- Внутри перечня информация о нежелательных реакциях приводится по системам органов
- При большом количестве реакций структурные перечни целесообразно вынести в приложение к ПОБЛП
- В разделе "Описание реакции" в начале указывается наиболее тяжелое состояние, наблюдавшееся у пациента в ходе ее развития (например, угроза жизни, госпитализация, даже если реакция закончилась полным выздоровлением)
- В разделе "Комментарий" может быть приведена позиция держателя регистрационного удостоверения относительно влияния на соотношение пользы и риска применения препарата (например, производитель не согласен с оценкой сообщившего о нежелательной реакции; имелось сопутствующее лечение или лекарственное взаимодействие, которое могло привести к развитию осложнений, лекарственное средство применялось вне показаний, указанных в инструкции лекарственного препарата)
- Если у пациента в одно время выявлено несколько симптомов нежелательной реакции, данное наблюдение классифицируется как один эпизод. Тяжесть реакции при этом оценивается по наиболее тяжелому симптому
- Если же у одного пациента выявлено несколько нежелательных реакций, разделенных временным промежутком, каждая из таких реакций приводится в структурированном перечне самостоятельно
- В некоторых случаях держатель регистрационного удостоверения может счесть целесообразным подготовить отдельные структурированные перечни для каждой из дозировок лекарственных форм или показаний к использованию препарата (например, в случае значительного различия показаний)
- При малом количестве случаев раздел может быть заменен обобщенной характеристикой в виде текстового изложения данных реакций
Пример заполнения структурированного перечня дан в приложении 6.
Обобщенные таблицы
В целях обеспечения возможности количественного анализа в ПОБЛП рекомендуется представлять информацию о количестве выявленных нежелательных реакций со стороны различных органов и систем организма.
Данные сведения целесообразно представлять в виде обобщенной таблицы (см. таблица 4).
Таблица 4. Рекомендуемый формат обобщенной таблицы нежелательных реакций на лекарственный препарат.
- Для реакций, информация о которых получена держателем регистрационного удостоверения от специалистов здравоохранения, органов управления здравоохранением Российской Федерации либо из литературных источников, приводятся количественные показатели следующих типов реакций:
- серьезные предвиденные (С/П)
- серьезные непредвиденные (С/НП)
- несерьезные предвиденные (Н/П)
- несерьезные непредвиденные (Н/НП)
- Для реакций, выявленных в ходе клинических исследований, спонсором которых является держатель регистрационного удостоверения лекарственного препарата, целесообразно представить информацию о серьезных предвиденных и серьезных непредвиденных реакциях.
- Внутри обобщенной таблицы реакции предпочтительно группировать сначала по системам органов, а затем по симптомам нежелательных реакций (WHO-ART <1>, MedDRA <2>).
--------------------------------
<1> http://www.umc-products.com/DynPage.aspx?id=4918
<2> http://www.meddramsso.com/
Пример заполнения обобщенной таблицы приведен в приложении 7.
Обобщенная таблица способов представления данных о нежелательных реакциях в зависимости от их предвиденности, серьезности и источника поступления информации представлена в таблице 5.
Таблица 5. Форма представления информации о нежелательных реакциях в зависимости от источника поступления, серьезности и предвиденности.
3.9. Сведения о безопасности лекарственных препаратов, полученные в ходе исследований
В данном разделе целесообразно представить информацию обо всех завершенных исследованиях (доклинических, клинических, эпидемиологических), в которых получена новая информация по безопасности лекарственного препарата.
Рекомендуется привести данные как о завершенных, так и продолжающихся исследованиях, организованных держателем регистрационного удостоверения в отчетный период. Также в раздел включаются данные из литературных источников, касающиеся сведений о безопасности лекарственных препаратов соответствующего МНН.
В разделе рекомендуется предусмотреть следующие части:
- Впервые анализируемые исследования, спонсором которых являлся держатель регистрационного удостоверения. Информация о доклинических, клинических, эпидемиологических исследованиях должна включать в себя наименование протокола исследования, краткую информацию о протоколе (например, сведения о цели и дизайне исследования), дату его начала, дату окончания, количество участников. Должна быть приведена характеристика проблемы безопасности, выявленной в ходе исследования. Сведения о завершенных исследованиях могут включать описание результатов с позиций влияния этих данных на соотношение пользы и риска применения препарата. Для продолжающихся исследований либо для завершенных исследований, анализ результатов которых не завершен, целесообразно представить результаты промежуточного анализа.
- Сведения о новых исследованиях лекарственного препарата, завершенных или начатых держателем регистрационного удостоверения лекарственного препарата в течение отчетного периода, в ходе которых выявлена значимая информация по безопасности лекарственного препарата.
- Сведения о специальных исследованиях безопасности (доклинических, клинических, эпидемиологических и др.), завершенных или начатых держателем регистрационного удостоверения лекарственного препарата в течение отчетного периода. Информация приводится в порядке, описанном в предыдущем пункте.
- Публикации об исследованиях препаратов данного международного непатентованного названия, в которых выявлена значимая информация по безопасности. Сведения о публикации целесообразно привести в объеме резюме научной статьи с указанием библиографических данных источника. При необходимости копии публикаций могут быть приложены к ПОБЛП.
3.10. Дополнительная информация по безопасности
Информация, связанная с эффективностью
В случае если лекарственный препарат используется для лечения серьезных, представляющих угрозу для жизни заболеваний, следует привести описание и обоснование соответствующих сообщений об отсутствии эффективности, подтвержденные специалистами здравоохранения (в форме аннотированных эпизодов реакций или структурированных перечней).
Важная информация, полученная после окончания периода сбора
данных для ПОБЛП
В данном разделе приводится любая новая важная информация, полученная после окончания периода сбора данных для подготовки ПОБЛП.
Например, в раздел могут быть включены сведения о недавних решениях зарубежных регуляторных агентств по ограничению обращения лекарственных препаратов данного международного непатентованного названия, недавно полученных спонтанных сообщений о значимых нежелательных реакциях и прочие новые данные, влияющие на соотношение пользы и риска применения лекарственного препарата.
Сведения, полученные после окончания сбора данных по безопасности лекарственного препарата для данного отчета, следует тем не менее принять во внимание при подготовке раздела "Итоговая оценка безопасности лекарственного препарата".
3.11. Итоговая оценка безопасности лекарственного препарата
Итоговая оценка профиля безопасности лекарственного препарата должна включать краткий, но всесторонний анализ представленных в ПОБЛП данных о нежелательных реакциях и проблемах безопасности. Необходимо принимать во внимание любую важную информацию, полученную как в период сбора данных по безопасности для данного отчета, так и в период написания ПОБЛП.
В ходе итоговой оценки профиля безопасности лекарственного препарата рекомендуется отразить следующие аспекты его применения (при наличии):
- Изменение характеристик нежелательных реакций, описанных в инструкции по медицинскому применению препарата, включая частоту развития реакций, тяжесть их симптомов и исходов, изменения характеристики групп пациентов, у которых они наблюдались
- Сведения о серьезных непредвиденных реакциях (проводится комплексная оценка данных о серьезных непредвиденных реакциях, включая сведения из предыдущих ПОБЛП)
- Новые данные по лекарственным взаимодействиям
- Данные о передозировках лекарственного препарата (случайных или преднамеренных)
- Сведения о возможных нежелательных реакциях при применении лекарственного препарата вне показаний, указанных в инструкции к применению
- Информация о случаях возникновения зависимости при применении лекарственных препаратов, оценка эффективности мер, предпринимавшихся для терапии передозировок
- Ошибки применения препарата
- Использование лекарственных препаратов в нелегальных целях
- Данные о применении препарата при беременности и лактации (положительные и отрицательные)
- Новые данные о применении лекарственных препаратов у отдельных категорий пациентов, в том числе у пожилых людей, детей, женщин в период беременности и лактации, пациентов с почечной и печеночной недостаточностью и других групп в зависимости от сферы применения препарата
- Новые данные о безопасности длительного применения лекарственных препаратов
В каждом из приводимых пунктов можно привести оценку влияния полученной информации на соотношение пользы и риска применения лекарственного препарата, а также позицию держателя регистрационного удостоверения относительно целесообразности внесения изменений в инструкцию по медицинскому применению лекарственного препарата либо иных действий, направленных на снижение риска его применения (например, изменение порядка отпуска препарата из аптек или публикации информационного письма держателя регистрационного удостоверения лекарственного средства, адресованного специалистам здравоохранения).
Анализ рекомендуется завершить перечнем проблем безопасности, требующих дополнительного мониторинга, регуляторных решений или иных мероприятий, направленных на повышение безопасности использования лекарственного препарата.
Кроме того, держатель регистрационного удостоверения может оценить эффективность мер, предпринимавшихся специалистами здравоохранения для лечения выявленных нежелательных реакций и передозировок, а также возможные ошибки в диагностике нежелательных реакций лекарственного препарата. Данная информация может служить основой оценки необходимости дополнительного информирования медицинской общественности по вопросам безопасного использования лекарственного препарата.
3.12. Заключение
В заключение ПОБЛП рекомендуется привести следующие сведения:
- информацию о возможном изменении соотношения пользы и риска применения лекарственного препарата за отчетный период
- информацию об основных проблемах безопасности лекарственного препарата, впервые выявленных или сохранявшихся в период сбора данных ПОБЛП
- информацию о необходимости изменения инструкции по медицинскому применению лекарственного препарата либо иных мероприятиях, направленных на обеспечение безопасности его применения.
4. Представление ПОБЛП в Федеральную службу по надзору в сфере здравоохранения
ПОБЛП предоставляются в Росздравнадзор держателем регистрационного удостоверения или другой организацией, которая уполномочена держателем регистрационного удостоверения в соответствии с контрактным соглашением (или уполномоченным представителем держателя регистрационного удостоверения). При предоставлении ПОБЛП уполномоченным представителем держателя регистрационного удостоверения в направительном письме должны быть указаны контрактные отношения (производство по лицензии, лицензионное соглашение по дистрибуции и др. виды контрактных отношений).
При представлении зарубежными заявителями государственной регистрации ПОБЛП в форматах, описанных руководствами ICH E2C (R1) и ICH E2C (R2), в случае, если подготовка ПОБЛП с использованием международных данных по безопасности лекарственного препарата осуществляется в сроки, установленные законодательством ЕС и США на основании положений ICH E2C (R1) (60 дней с момента окончания периода сбора данных для очередного ПОБЛП), держателю регистрационного удостоверения данного лекарственного препарата или его уполномоченному лицу следует не менее чем за 60 дней до срока представления периодического отчета, установленного приказом Минздравсоцразвития России от 26.08.2010 N 757н, проинформировать Росздравнадзор о задержке представления ПОБЛП.
При этом Росздравнадзор может запросить у держателя регистрационного удостоверения в срок, установленный для подачи ПОБЛП, в соответствии с приказом Минздравсоцразвития России от 26.08.2010 N 757н представить в Федеральную службу по надзору в сфере здравоохранения перечень нежелательных реакций на лекарственный препарат, выявленных держателем регистрационного удостоверения в Российской Федерации за отчетный период. Данный перечень рекомендуется представлять в Росздравнадзор в виде структурированного перечня, описанного в разделе 3.8 настоящих рекомендаций и в разделе 2.6.2 рекомендаций ICH E2C (R1) ("Line Listings").
ПОБЛП предоставляется в Федеральную службу по надзору в сфере здравоохранения на цифровом носителе (предпочтительно компакт-диск CD/DVD-R) с сопроводительным письмом.
К ПОБЛП рекомендуется прилагать версию последней инструкции по применению и представленные в Министерство здравоохранения Российской Федерации на момент подачи ПОБЛП проекты изменений в инструкцию по медицинскому применению (при наличии).
Также к ПОБЛП рекомендуется прилагать копии научных публикаций и сведений о зарубежных регуляторных решениях, связанных с безопасностью лекарственного препарата (см. выше).
При направлении объемных ПОБЛП, ПОБЛП со множеством приложений или нескольких ПОБЛП возможна запись ПОБЛП и прилагающихся к ним материалов на один электронный носитель с одним направительным письмом, в котором перечислены все предоставляемые ПОБЛП. При этом резюме ПОБЛП следует представлять в бумажном виде.
Сопроводительные письма направляются на имя руководителя Федеральной службы по надзору в сфере здравоохранения по адресу: 109074, Москва, Славянская пл., д. 4, стр. 1.
Дополнительно к описанному порядку электронная версия ПОБЛП с приложениями и направительным письмом может быть загружена держателями регистрационных удостоверений через индивидуальный кабинет информационного ресурса "Фармаконадзор" АИС Росздравнадзора.
5. Дополнительная литература:
- Астахова А.В., Лепахин В.К. "Лекарства: неблагоприятные побочные реакции и контроль безопасности". Москва, 2008.
- Викторов А.П., Мальцев В.И., Белоусов Ю.Б. "Безопасность лекарств". Руководство по фармаконадзору. Киев, Морион (Украина), 2007 год.
- Стефанов А.В., Бахтиарова Т.А., Варченко В.Г. и др. "Фармацевтический сектор: фармаконадзор за лекарственными препаратами для человека", Морион (Украина), (перевод тома 9A Руководства Европейской Комиссии по регулированию лекарственных средств в Европейском Союзе), Морион (Украина).
- Н.В. Юргель, В.Г. Кукес, "Профилактика неблагоприятных побочных реакций. Врачебная тактикарационального выбора и применения лекарственных средств". Москва, 2009.
- Руководства Международной Конференции по Гармонизации (http://www.ich.org/products/guidelines/efficacy/article/efficacy-guidelines.html:
- E2A. Управление данными по клинической безопасности: определения и стандарты экстренной отчетности
- E2B (R3): Управление данными клинической безопасности: элементы данных для передачи сообщений об индивидуальных случаях, связанных с безопасностью
- E2C (R1): Управление данными клинической безопасности: Периодическая отчетность о безопасности зарегистрированных лекарственных препаратов
- E2C (R3): Периодический отчет о соотношении пользы и риска [применения лекарственного препарата] (PBER)
- E2D: Управление данными пострегистрационными данными безопасности: определения и стандарты экстренной отчетности
- E2E: Планирование фармаконадзора
- E2F: Отчет по безопасности исследуемого лекарственного средств
- Всемирная организация здравоохранения "Введение в исследования потребления лекарственных средств" (WHO) IntroductiontoDrugUtilizationResearch, 2003
- Руководство Европейского Агентства по Лекарственным Средствам (ЕМА) по надлежащей практике фармаконадзора (Good pharmacovigilance practice emodules) (http://www.ema.europa.eu/ema/index.jsp?cure=pages/regulation/document_listing/document_listing_000345.jsp&mid=WC0b01ac058058f32c)
- Module I: Pharmacovigilance systems and their quality systems;
- Module II: Pharmacovigilance systems master files;
- Module V: Risk management systems;
- Module VI: Management and reporting of adverse reactions to medicinal products;
- Module VII: Periodic safety update reports;
- Module VIII: Post-authorization safety studies;
- Module IX: Signal Management
- Good Pharmacovigilance Practice Guide, (2008). Pharmaceutical Press.
- Orleans-Lindsay, J. (2012). Pharmacovigilance Medical Writing: A Good Practice Guide, Wiley.
Приложения
к методическим рекомендациям
по подготовке разработчиками
и производителями лекарственных
препаратов, находящихся в обращении
на территории Российской Федерации,
периодических отчетов по безопасности
лекарственных препаратов.
Приложение 1
ИСТОЧНИКИ
ИНФОРМАЦИИ ПО РЕГУЛЯТОРНЫМ РЕШЕНИЯМ В ОБЛАСТИ БЕЗОПАСНОСТИ
ЛЕКАРСТВЕННЫХ СРЕДСТВ (НА РУССКОМ И АНГЛИЙСКОМ ЯЗЫКАХ,
ССЫЛКИ ДЕЙСТВИТЕЛЬНЫ ПО СОСТОЯНИЮ НА СЕНТЯБРЬ 2012 ГОДА)
- Бюллетень ВОЗ по лекарственным средствам http://www.who.int/medicines/publications/newsletter/en/
- Журнал "Безопасность лекарств и фармаконадзор" http://www.regmed.ru/fnz/blf.aspx
- Сайт Европейского Медицинского Агентства http://www.ema.europa.eu/
- Документы Рабочей группы по фармаконадзору Европейского Медицинского Агентства http://www.ema.europa.eu/ema/index.jsp?curl=pages/news_and_events/document_ listing/document_listing_000198.jsp&mid=WC0b01ac0580033aal
- Сайт Управления по контролю за продуктами питания и лекарственными средствами США http://www.fda.gov/Drugs/DrugSafety/default.htm
- Сайт Регуляторного агентства Великобритании по медицинской продукции http:http://www.mhra.gov.uk/Safetyinformation/Safetywarningsalertsandrecalls/Safetywarningsandmessagesformedicines/index.htm
Приложение 2
ПРИМЕР
ЗАПОЛНЕНИЯ ТАБЛИЦЫ С ИНФОРМАЦИЕЙ О РЕГИСТРАЦИИ
ЛЕКАРСТВЕННОГО ПРЕПАРАТА
Приложение 3
ОБЩАЯ ИНФОРМАЦИЯ
ОБ УСТАНОВЛЕННОЙ СУТОЧНОЙ ДОЗЕ И ОСНОВАННОЙ НА НЕЙ МЕТОДИКЕ
ПОДСЧЕТА ПОТРЕБЛЕНИЯ ЛЕКАРСТВЕННОГО СРЕДСТВА
Установленная суточная доза ЛС (DDD - Defined Daily Dose) является расчетной средней поддерживающей суточной дозой лекарственного средства, применяемого по основному показанию у взрослых массой 70 кг. Установленная суточная доза ЛС (DDD) - это техническая единица измерения, которая не аналогична рекомендуемой суточной дозе, зависящей от степени тяжести, характера течения заболевания, массы тела пациента и т.д. Она определяется для тех лекарственных препаратов, которым присвоен код Анатомо-терапевтической классификации АТХ.
Необходимо помнить, что DDD может быть определена только для наиболее широко используемых комбинированных лекарственных средств.
DDD препаратов, включенных в АТХ, приведены на сайте Сотрудничающего центра ВОЗ по методологии фармацевтической статистики (см. http://www.whocc.no/ddd/definition_and_general_considera/ и http://www.whocc.no/atc_ddd_index/)
Подсчет объемов потребления лекарственного препарата в пациенто-годах на основе установленной суточной дозы определяется по формуле
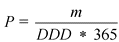
где
P - пациенто-годы
m - общая масса действующего вещества, содержавшегося в реализованных упаковках препарата (г)
Y - количество дней в году (365)
DDD - установленная суточная доза (г)
Дополнительно к количеству пациенто-лет в ПОБЛП можно привести примерные данные о количестве пациентов, которые могли получить стандартный курс лечения препаратом за отчетный период.
Этот показатель рассчитывается по формуле
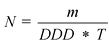
где
N - число стандартных курсов лечения
m - общее количество действующего вещества, содержавшегося в реализованных упаковках препарата (г)
T - длительность стандартного курса лечения
DDD - установленная суточная доза (г)
Для препаратов, не имеющих установленной суточной дозы, объем потребления можно привести в количестве единиц дозирования препарата, использованных за отчетный период, количестве выписанных рецептов, а при отсутствии более точных сведений - в общем объеме действующего вещества.
Пример расчета объемов потребления
1. В клинических исследованиях препарат применялся у ... пациентов
(указывается количество пациентов, у которых использовался препарат в ходе клинических исследований в течение отчетного периода как интервенционных, так и наблюдательных. Также могут быть указаны данные регистров препарата или заболевания (при наличии). Учитываются данные из завершенных двойных слепых и наблюдательных исследований. Данные о применении препарата в продолжающихся двойных слепых исследованиях указываются приблизительно (примерное количество человек, получающих исследуемое ЛС, в доле от общего числа включенных в исследование пациентов).
2. Данные о потреблении препарата на фармацевтическом рынке подсчитываются на основании установленной суточной дозы.
Препарат NN находится в обращении в двух лекарственных формах
- таблетки 100 мг
- раствор для внутримышечного введения 50 мг/мл
Курс лечения по основному показанию препаратом составляет 7 дней (выбирается показание, по которому лекарственный препарат наиболее часто применяется в медицинской практике).
Таблетки выпускаются в упаковках - по 10 и по 50 таблеток в упаковке.
1 ампула содержит 2 мл раствора препарата NN. Ампулы реализуются по 10 ампул в упаковке.
За отчетный период производителем реализовано
- 3 млн упаковок таблеток NN по 10 таблеток в упаковке
- 1 млн упаковок таблеток NN по 10 таблеток в упаковке
- 500 000 упаковок ампул NN
Стандартный курс лечения препаратом по основному показанию составляет 10 дней.
Методика рассчета объема потребления препарата в пациенто-годах
1. Определяется общая масса действующего вещества во всех реализованных упаковках лекарственного препарата
- 3 000 0000 уп. * 10 (упаковок в пачке) * 0,1 (масса активного в-ва в граммах в 1 таб.) = 3 000 000 г действующего в-ва
- 1 000 0000 уп. * 50 (упаковок в пачке) * 0,1 (г) =
- = 5 000 000 г действующего в-ва
- 500 000 * 10 (ампул в пачке) * 0,1 (масса в-ва в одной ампуле) = 500 000 г действующего
Итого: за отчетный период реализовано 8 500 000 г (8500 кг) действующего вещества
2. Определение установленной суточной дозы действующего вещества
Установленную суточную дозу можно найти на сайте Сотрудничающего центра ВОЗ по методологии фармацевтической статистики
http://www.whocc.no/atc_ddd_index/ повтор
Предположим, что установленная суточная доза для действующего вещества препарата NN составляет 0,2 г
3. Подсчет количества пациенто-лет проводится по формуле:
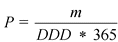
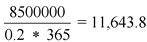
Таким образом, экспозиция препарата за отчетный период составляет = 11 644 пациенто-лет
3. Количество стандартных курсов лечения препаратом за отчетный период определяется по формуле:
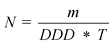
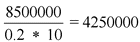 стандартных курсов лечения
стандартных курсов лечения
Приложение 4
ПРИМЕРНАЯ СТРУКТУРА
РАЗДЕЛА ПОБЛП "ОТДЕЛЬНЫЕ ЭПИЗОДЫ НЕЖЕЛАТЕЛЬНЫХ РЕАКЦИЙ,
ИМЕЮЩИЕ КЛИНИЧЕСКУЮ ЗНАЧИМОСТЬ"
Примерная структура раздела
Важно: В начале каждого пункта рекомендуется дать краткую обобщенную характеристику описываемых реакций.
Приложение 5
ПРИМЕР ОПИСАНИЯ ОДНОЙ НЕЖЕЛАТЕЛЬНОЙ РЕАКЦИИ
Сообщение поступило от специалиста здравоохранения. У 55-летней пациентки с диагнозом ... и почечной недостаточностью в анамнезе через три дня после начала терапии лекарственным препаратом N развилась фибрилляция предсердий. Одновременно принимала лекарственные препараты ... [указываются даты начала и окончания сопутствующей терапии, если известно]. Пациентка была госпитализирована по поводу фибрилляции. Терапия лекарственным препаратом N была прекращена. По поводу фибрилляции предсердий назначались ... . Спустя ... дней после госпитализации пациентке был имплантирован кардиостимулятор. Росздравнадзор проинформирован о данной нежелательной реакции извещением (N, дата, при необходимости указываются номера дополнительных извещений АИС Росздравнадзора). Пациентка была выписана из больницы с улучшением.
Вследствие госпитализации, связанной с фибрилляцией предсердий, случай квалифицирован как серьезная нежелательная реакция.
Комментарий держателя регистрационного удостоверения: Согласно инструкции по применению фибрилляция предсердий является очень редкой нежелательной реакцией на лекарственный препарат N. Лекарственный препарат X, применявшийся одновременно с лекарственным препаратом N также способен вызывать фибрилляцию предсердий. Увеличения частоты риска фибрилляции предсердий у больных с ишемической болезнью сердца на фоне совместного приема лекарственных препаратов N и X описано в инструкциях обоих препаратов. Причинно-следственная связь данной нежелательной реакции с применением лекарственного препарата N классифицирована как возможная.
Приложение 6
ПРИМЕР ЗАПОЛНЕНИЯ СТРУКТУРИРОВАННОГО ПЕРЕЧНЯ
Серьезные реакции, информация о которых поступила напрямую держателю регистрационного удостоверения от специалистов здравоохранения
Приложение 7
ПРИМЕР ЗАПОЛНЕНИЯ ОБОБЩЕННОЙ ТАБЛИЦЫ