Рекомендация Коллегии Евразийской экономической комиссии от 16.01.2018 N 2
КОЛЛЕГИЯ ЕВРАЗИЙСКОЙ ЭКОНОМИЧЕСКОЙ КОМИССИИ
РЕКОМЕНДАЦИЯ
от 16 января 2018 г. N 2
О РУКОВОДСТВЕ
ПО КАЧЕСТВУ ЛЕКАРСТВЕННЫХ ПРЕПАРАТОВ С МОДИФИЦИРОВАННЫМ
ВЫСВОБОЖДЕНИЕМ ДЛЯ ПРИЕМА ВНУТРЬ
Коллегия Евразийской экономической комиссии в соответствии со статьей 30 Договора о Евразийском экономическом союзе от 29 мая 2014 года, пунктом 3 статьи 3 Соглашения о единых принципах и правилах обращения лекарственных средств в рамках Евразийского экономического союза от 23 декабря 2014 года,
в целях устранения различий в требованиях, предъявляемых к подтверждению качества при внесении изменений в регистрационное досье и оценке эквивалентности лекарственных препаратов с модифицированным высвобождением для приема внутрь, установленных законодательством государств - членов Евразийского экономического союза,
рекомендует государствам - членам Евразийского экономического союза по истечении 6 месяцев с даты опубликования настоящей Рекомендации на официальном сайте Евразийского экономического союза при планировании проведения исследований биоэквивалентности в соответствии с Правилами проведения исследований биоэквивалентности лекарственных препаратов в рамках Евразийского экономического союза, утвержденными Решением Совета Евразийской экономической комиссии от 3 ноября 2016 г. N 85, и при внесении изменений в регистрационные досье лекарственных препаратов с модифицированным высвобождением для приема внутрь в соответствии с Правилами регистрации и экспертизы лекарственных средств для медицинского применения, утвержденными Решением Совета Евразийской экономической комиссии от 3 ноября 2016 г. N 78, применять Руководство по качеству лекарственных препаратов с модифицированным высвобождением для приема внутрь согласно приложению.
Врио Председателя Коллегии
Евразийской экономической комиссии
К.МИНАСЯН
Приложение
к Рекомендации Коллегии
Евразийской экономической комиссии
от 16 января 2018 г. N 2
РУКОВОДСТВО
ПО КАЧЕСТВУ ЛЕКАРСТВЕННЫХ ПРЕПАРАТОВ С МОДИФИЦИРОВАННЫМ
ВЫСВОБОЖДЕНИЕМ ДЛЯ ПРИЕМА ВНУТРЬ
I. Общие положения
1. Настоящее Руководство определяет требования к качеству лекарственных препаратов с модифицированным высвобождением для приема внутрь.
2. Настоящее Руководство распространяется на лекарственные формы, в которых определенным образом осуществлено изменение (модификация) скорости и (или) места высвобождения действующего вещества (действующих веществ) в сравнении с лекарственными формами со стандартным высвобождением. Такая модификация может проводиться для продления терапевтической активности лекарственного препарата, уменьшения токсического действия, защиты действующего вещества от деградации вследствие низкого значения pH, высвобождения действующего вещества из лекарственной формы в заданном сегменте желудочно-кишечного тракта для оказания местного действия или в определенные временные точки, а также в других целях.
3. Настоящее Руководство охватывает различные части регистрационного досье, связанные с качеством лекарственного препарата, и их следует рассматривать вместе с соответствующими актами, входящими в право Евразийского экономического союза (далее - Союз), в части клинических аспектов исследований.
4. Настоящее Руководство рекомендуется применять при подаче заявления о регистрации лекарственного препарата в уполномоченный орган государства - члена Союза (далее - государство-член) при проведении экспертизы регистрационных досье, а также при разработке лекарственных препаратов. Настоящее Руководство применяется при планировании и проведении научных исследований по фармацевтической разработке, а также при составлении регистрационных досье.
5. Введение в действие настоящего Руководства не влечет за собой новых требований к зарегистрированным лекарственным препаратам.
II. Определения
6. Для целей настоящего Руководства используются понятия, которые означают следующее:
"биосерия" (biobatch) - серия, используемая в клиническом исследовании биодоступности (биоэквивалентности) или клиническом исследовании эффективности (подтверждающем наличие функциональных характеристик лекарственной формы). Размер биосерии соответствует как минимум размеру опытно-промышленной серии, то есть для твердых лекарственных форм для приема внутрь он составляет не менее 10% от размера серии при полномасштабном производстве или 100 000 единиц лекарственной формы (в зависимости от того, какой из этих показателей больше);
"внешняя прогностическая способность" (external predictability) - прогностическая способность, в отношении которой проводится оценка с использованием результатов, отличных от тех, на основании которых установлена корреляция "in vivo - in vitro" (насколько точно модель прогнозирует результаты);
"внутренняя прогностическая способность" (internal predictability) - прогностическая способность, в отношении которой проводится оценка с использованием результатов исходных испытаний, на основании которых установлена корреляция "in vivo - in vitro" (насколько точно модель описывает результаты, использованные для установления корреляции "in vivo - in vitro");
"вспомогательное вещество, контролирующее высвобождение" (release controlling excipient) - вспомогательное вещество с определяющим влиянием на высвобождение действующего вещества;
"высвобождение нулевого порядка" (zero order release) - высвобождение действующего вещества, скорость которого не зависит от времени;
"деконволюция" (обратная свертка) (deconvolution) - определение кинетики поступления действующего вещества в организм (как правило, по абсорбции или растворению in vivo) с помощью математической модели, основанной на интеграле конволюции (свертка функций). Например, зависимость скорости абсорбции (rabs) от времени, которая приводит к концентрации действующего вещества в плазме (c(t)), может быть рассчитана путем решения следующего интеграла свертки для rabs:
 ,
,
где:
![]() - функция, отражающая зависимость концентрации действующего вещества от времени, получаемую при мгновенной абсорбции единичного количества действующего вещества и обычно рассчитываемую по результатам внутривенного струйного (болюсного) введения, приема раствора, суспензии или быстро высвобождающихся лекарственных форм с немедленным высвобождением для приема внутрь;
- функция, отражающая зависимость концентрации действующего вещества от времени, получаемую при мгновенной абсорбции единичного количества действующего вещества и обычно рассчитываемую по результатам внутривенного струйного (болюсного) введения, приема раствора, суспензии или быстро высвобождающихся лекарственных форм с немедленным высвобождением для приема внутрь;
t - время;
rabs - скорость абсорбции;
u - переменная интегрирования;
"демпинг дозы" (сброс дозы) (dose dumping) - непреднамеренно быстрое высвобождение действующего вещества из лекарственной формы;
"конволюция" (свертка) (convolution) - прогнозирование концентрации действующего вещества в плазме с помощью математической модели, основанной на интеграле конволюции (свертка функций). Например, для прогнозирования концентрации действующего вещества в плазме (c(t)) исходя из зависимости скорости абсорбции (rabs) от времени может быть использован следующий интеграл свертки:
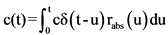 ,
,
где:
![]() - функция, отражающая зависимость концентрации действующего вещества от времени, получаемую при мгновенной абсорбции единичного количества действующего вещества и обычно рассчитываемую по результатам внутривенного струйного (болюсного) введения;
- функция, отражающая зависимость концентрации действующего вещества от времени, получаемую при мгновенной абсорбции единичного количества действующего вещества и обычно рассчитываемую по результатам внутривенного струйного (болюсного) введения;
t - время;
rabs - скорость абсорбции;
u - переменная интегрирования;
"корреляция in vivo - in vitro" (in vivo - in vitro correlation) - вероятностная зависимость (взаимосвязь) линейного характера параметров биодоступности лекарственного препарата от его физико-химических свойств или характеристик. Прогностическая математическая модель, описывающая зависимость между in vitro свойством лекарственной формы с пролонгированным высвобождением (как правило, скоростью или степенью растворения либо высвобождением действующего вещества) и соответствующим in vivo ответом, например, плазменной концентрацией действующего вещества или его абсорбированным количеством;
"лекарственные формы с модифицированным высвобождением" (modified release dosage forms) - лекарственные формы, скорость и (или) место высвобождения действующего (действующих) вещества (веществ) которых отличаются от лекарственных форм со стандартным высвобождением при том же пути введения. Модификация достигается путем разработки специального состава и (или) специальной технологии производства. К лекарственным формам с модифицированным высвобождением относятся лекарственные формы с пролонгированным, отсроченным (отложенным), пульсирующим и ускоренным высвобождением;
"лекарственные формы с пролонгированным высвобождением", "лекарственная форма с замедленным высвобождением" (prolonged release dosage forms) - лекарственные формы с модифицированным высвобождением, характеризующиеся более медленным высвобождением, чем у лекарственных форм со стандартным высвобождением при том же пути введения. Пролонгированное высвобождение достигается путем разработки специального состава и (или) специальной технологии производства;
"лекарственная форма со стандартным высвобождением", "лекарственная форма с немедленным высвобождением" (conventional release dosage form) - лекарственная форма с высвобождением действующего вещества, преднамеренно не модифицированным с помощью специального состава и (или) специальной технологии производства. Профиль растворения действующего вещества твердых лекарственных форм существенно зависит от внутренних свойств этого вещества;
"погрешность прогнозирования" (percent prediction error) - выраженная в процентах погрешность прогнозирования концентрации действующего вещества, которая рассчитывается по следующей формуле:
 ;
;
"серии с предельным содержанием" (side batch) - серии, соответствующие предполагаемому верхнему или нижнему пределу спецификации высвобождения in vitro, которая составлена на основании описанного производственного процесса путем установления его параметров в диапазоне максимальной вариабельности, ожидаемой по результатам исследований по валидации процесса;
"среднее время абсорбции" (mean absorption time) - время достижения действующим веществом системного кровотока с момента применения лекарственного препарата, равное среднему времени процессов высвобождения и абсорбции in vivo ввиду их протекания во входном компартменте (камере):
MAT = MRToral - MRTi.v.,
где:
MRToral - среднее время удерживания действующего вещества при приеме внутрь;
MRTi.v. - среднее время удерживания действующего вещества при внутривенном введении;
"среднее время растворения in vitro" (mean in vitro dissolution time) - среднее время растворения лекарственного препарата in vitro:
 ,
,
где:
MDTvitro - среднее время растворения лекарственного препарата в модельном растворе;
M(t) - количество действующего вещества лекарственного препарата, перешедшее в раствор к определенному моменту времени t;
![]() - количество действующего вещества лекарственного препарата, переходящее в раствор при условии экстраполирования времени наблюдения в бесконечность;
- количество действующего вещества лекарственного препарата, переходящее в раствор при условии экстраполирования времени наблюдения в бесконечность;
"среднее время растворения in vivo" (mean in vivo dissolution time) - среднее время растворения лекарственного препарата in vivo, которое рассчитывается по следующей формуле:
MDTsolid = MRTsolid - MRTsolution,
где:
MRTsolid - среднее время удерживания действующего вещества при приеме твердой лекарственной формы;
MRTsolution - среднее время удерживания действующего вещества при введении раствора;
"среднее время удерживания in vivo" (mean in vivo residence time) - среднее время удерживания действующего вещества в организме, которое рассчитывается по следующей формуле:
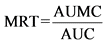 ,
,
где:
AUC (area under the curve) - полная площадь под фармакокинетической кривой "концентрация - время";
AUMC (area under the moment curve) - полная площадь под кривой первого момента "произведение времени на концентрацию - время";
"статистические моменты" (statistical moments) - параметры, описывающие характеристики зависимости концентрации действующего вещества в плазме от времени (площадь, среднее время удерживания и дисперсия среднего времени удерживания) и скорость экскреции его с мочой;
"условия достаточного разбавления" (sink conditions) - условия, при которых количество вещества в растворе при завершении испытания на растворение не превышает 30% от концентрации его в насыщенном растворе.
III. Область применения
7. В настоящем Руководстве устанавливаются требования к качеству лекарственных форм с модифицированным высвобождением для приема внутрь, в частности требования к фармацевтической разработке и проведению испытаний in vitro. Настоящее Руководство охватывает лекарственные формы для приема внутрь только с пролонгированным высвобождением и отсроченным (отложенным) высвобождением, основанным на принципе гастрорезистентности (устойчивости к действию желудочного сока). Лекарственные формы с пульсирующим и ускоренным высвобождением не входят в область применения настоящего Руководства. Лекарственные формы с отсроченным (отложенным) высвобождением, разработанные на основе других принципов, в том числе с высвобождением в определенной области желудочно-кишечного тракта, под влиянием определенного триггер-фактора (например, ферментов) или в определенное время после приема внутрь отдельно не рассматриваются.
8. Положения настоящего Руководства в отношении лекарственных форм с пролонгированным высвобождением для приема внутрь распространяются на другие лекарственные формы с модифицированным высвобождением, предназначенные для приема внутрь или другого пути введения.
IV. Лекарственные формы для приема внутрь
с пролонгированным высвобождением
1. Фармацевтическая разработка
Основные положения
9. Качество лекарственных форм с пролонгированным высвобождением непрерывно совершенствуется в процессе разработки нового лекарственного препарата. Подбор состава лекарственного препарата проводится при разработке на сериях небольшого масштаба с учетом физико-химических свойств действующего вещества, его стабильности и характеристик абсорбции в желудочно-кишечном тракте. После подбора компонентов состава лекарственного препарата начинается последовательное масштабирование производственного процесса. В течение указанного времени предполагается выполнение корректирующих действий, необходимых для осуществления полномасштабного производства. Такие корректирующие действия могут сводиться к изменению состава, производственных процессов, оборудования или производственной площадки.
10. В некоторых случаях корректирующие действия могут оказывать влияние на свойства лекарственного препарата. В связи с этим следует разработать испытание на растворение in vitro, способное обнаружить изменения, которые могут повлиять на эффективность или безопасность лекарственного препарата.
11. Фармацевтическая разработка должна устанавливать связь (качественную или количественную) между фармакокинетическими параметрами при высвобождении действующего вещества in vivo и скоростью растворения in vitro.
12. Состав лекарственной формы, подобранный при разработке, должен оцениваться в различных условиях растворения для определения его чувствительности (устойчивости) в ожидаемых окружающих физиологических условиях после введения. Дискриминационная способность условий проведения испытаний, выбранных для рутинного контроля, может определяться путем сравнения данных растворения in vitro и данных биодоступности различных составов. Рекомендуется устанавливать корреляцию in vivo - in vitro. При наличии корреляции in vivo - in vitro уровня A испытание на растворение после соответствующей валидации можно использовать в качестве квалифицирующего метода контроля, обладающего релевантностью in vivo, в то время как при отсутствии корреляции in vivo - in vitro уровня A испытание может использоваться только в качестве метода контроля качества.
13. Если коэффициент масштабирования превышает значение 10 (по сравнению с лабораторной (опытно-промышленной) биосерией), в целях верификации выбранных условий проведения испытания на растворение в части пригодности для выпуска клинического материала, масштабирования и производства по завершении масштабирования необходимо сравнить лабораторные (опытно-промышленные) серии с полномасштабными промышленными сериями в исследовании биодоступности.
Терапевтические цели и принцип функционирования
системы высвобождения
14. Необходимо указать терапевтические цели и обоснование создания лекарственной формы с пролонгированным высвобождением. Следует указать значимые для разработки лекарственного препарата фармакокинетические параметры (площадь под кривой "плазменная концентрация - время" с момента приема лекарственного препарата (AUC), максимальная плазменная концентрация (Cmax), время достижения максимальной плазменной концентрации (Tmax), период полувыведения из плазмы (![]() )) и физико-химические характеристики фармацевтической субстанции (растворимость при различных значениях pH, коэффициент распределения, размер частиц, полиморфизм).
)) и физико-химические характеристики фармацевтической субстанции (растворимость при различных значениях pH, коэффициент распределения, размер частиц, полиморфизм).
15. Необходимо представить подробную информацию о вспомогательном веществе (вспомогательных веществах), контролирующем (контролирующих) высвобождение, а также привести ссылки на нормативные документы по фармацевтической разработке.
16. Необходимо описать следующие характеристики системы пролонгированного высвобождения:
а) способ достижения пролонгированного высвобождения (тип мембраны, матрица и т.д.);
б) механизм и кинетика высвобождения (диффузия, эрозия, осмос и т.д. или их комбинации);
в) тип системы (например, нераспадающаяся цельная единица лекарственной формы, распадающаяся таблетка (капсула), содержащая гранулы (пеллеты), и т.д.).
17. Необходимо подтвердить, что препарат с пролонгированным высвобождением сохраняет характеристики высвобождения действующего вещества независимо от вариабельности физиологических условий. Такие изменения зависят, например, от времени транзита через желудок и кишечник, воздействия пищи, состава желудочного и кишечного сока при патологических состояниях и одновременного потребления алкоголя.
18. Лекарственные формы с пролонгированным высвобождением для приема внутрь не должны иметь разделительной риски (если только это не обосновано специальными исследованиями), так как разделение лекарственной формы с модифицированным высвобождением или другие манипуляции с лекарственными препаратами с модифицированным высвобождением могут отрицательно сказаться на характеристиках модифицированного высвобождения из лекарственной формы, что может привести к демпингу дозы. Любые рекомендации по разделению лекарственной формы с модифицированным высвобождением должны сопровождаться научным обоснованием об отсутствии влияния разделения на характеристики модифицированного высвобождения, в том числе результатами исследований in vitro и (или) in vivo.
Разработка методик испытания на растворение in vitro
19. Скорость высвобождения должна быть определена in vitro с помощью методики испытания на растворение. Разработка подходящей методики испытания должна быть основана на физико-химических характеристиках in vitro и in vivo характеристиках действующего вещества и лекарственного препарата с учетом механизма высвобождения.
20. Испытание на растворение in vitro должно:
устанавливать различия между сериями в зависимости от критических параметров процесса (CPP), которые могут оказывать влияние на требуемую биодоступность;
определять постоянство характеристик лекарственной формы лекарственного препарата от серии к серии (серий для опорных клинических испытаний, серий для исследований биодоступности и производственных серий);
определять стабильность соответствующих характеристик высвобождения лекарственного препарата в течение заявленного производителем срока хранения (срока годности) при заявленных условиях хранения.
В связи с этим следует проводить оценку in vitro лекарственной формы с пролонгированным высвобождением при различных условиях (средах растворения, pH (обычно в диапазоне pH 1,0 - 7,5, при необходимости до pH 8,0), типах приборов, перемешивании и т.д.). Необходимо определить условия проведения испытания, включая временные точки и частоту отбора проб, обеспечивающие наибольшую дискриминационную способность испытания.
21. Для обеспечения надлежащего контроля pH во время проведения испытания на растворение должен использоваться буферный раствор подходящей емкости. В противном случае может возникнуть необходимость контролирования pH среды в течение всего испытания. Если в среду растворения добавляется поверхностно-активное вещество, его выбор и количество необходимо обосновать. Необходимо обеспечить постоянство качества поверхностно-активного вещества между сериями.
22. Добавление ферментов в среду растворения представляется приемлемым, а в обоснованных производителем лекарственного препарата случаях даже целесообразным (например, при необходимости доставки лекарственного средства в толстую кишку посредством желатиновых капсул). При добавлении ферментов в среду растворения должны быть обоснованы их тип и концентрация. Кроме того, должно быть обеспечено постоянство качества ферментов от серии к серии, в том числе активности (МЕ/мг или МЕ/мл) или концентрации (мг/мл) соответственно. Указанная в Фармакопее Евразийского экономического союза (далее - Фармакопея Союза) концентрация фермента в искусственном желудочном соке (имитация желудочного сока), искусственном кишечном соке (имитация кишечного сока) значительно выше соответствующих физиологических значений. Необходимо использовать обоснованные концентрации ферментов, если они являются составной частью механизма контроля растворения. Использование биорелевантной среды может улучшить корреляцию с данными in vivo и обнаружить возможное влияние пищи.
23. Объем среды растворения предпочтительно должен обеспечить условия достаточного разбавления.
24. Для лекарственных форм, имеющих кинетику высвобождения нулевого порядка (с латентным периодом или без него), предпочтительно установить спецификацию скорости растворения (в виде значения количества действующего вещества в процентах заявленного количества этого действующего вещества в лекарственной форме, переходящего в раствор за 1 один час ("проценты в час") для заданного промежутка времени). В целях обоснования того, что лекарственную форму можно рассматривать как форму с кинетикой высвобождения нулевого порядка, должна быть дополнительно представлена графическая зависимость скорости растворения от времени растворения. Дополнительные подробные сведения относительно выбора прибора, условий испытания, валидации (квалификации) и критериев приемлемости содержатся в Фармакопее Союза.
25. Особое внимание следует уделять важности любого изменения свойств фармацевтической субстанции (например, размера частиц, полиморфизма), вспомогательных веществ, контролирующих высвобождение (например, размера частиц, гелеобразующих свойств), и процесса производства с точки зрения их влияния на биодоступность in vivo.
26. Методика количественного определения действующего вещества в пробах растворения должна быть валидирована в соответствии с актами, входящими в право Союза, с учетом стабильности действующего вещества, растворенного в среде, и влияния вспомогательных веществ.
27. Для различных дозировок одного и того же лекарственного препарата следует использовать идентичные или, если невозможно, сопоставимые условия проведения испытания.
28. В процессе разработки результаты испытаний на растворение in vitro для каждой единицы лекарственной формы, их среднее значение и мера вариабельности (например, стандартное отклонение или 95-процентный доверительный интервал) должны быть представлены для каждой временной точки. Использование других статистических подходов необходимо обосновать. Профиль растворения in vitro следует определять для всех дозировок и любых изменений состава и (или) процесса производства лекарственного препарата при его разработке.
Дискриминационная способность испытания на растворение
in vitro
29. Необходимо доказать, что испытание на растворение in vitro при выбранных условиях способно дискриминировать серии лекарственного препарата с приемлемыми и неприемлемыми характеристиками высвобождения.
30. Дискриминационная способность испытания на растворение in vitro может подтверждаться одним из следующих способов, указанных в порядке выбора их приоритетности:
а) включение в испытания на растворение in vitro серий лекарственного препарата, не показавших приемлемых фармакокинетических параметров in vivo. На основании результатов испытания могут быть составлены спецификации для отбраковки таких серий на основании данных растворения, что может быть количественно обосновано с помощью валидированной корреляции in vivo - in vitro, разработанной с учетом серий с неприемлемыми фармакокинетическими параметрами;
б) при отсутствии серий лекарственного препарата с неприемлемым профилем поведения in vivo - сравнением данных растворения со средними значениями фармакокинетического параметра (точечная оценка) в исследованиях фармакокинетики in vivo при помощи проверки рангового порядка результатов;
в) при невозможности осуществления применения положений подпунктов "а" и "б" настоящего пункта - целенаправленным изменением характеристики фармацевтической субстанции (например, изменением показателя распределения частиц по размерам), состава и (или) параметров производственного процесса для получения различных профилей растворения in vitro без получения данных испытания in vivo для тех же серий. Необходимо принять во внимание, что этот способ может привести к чрезмерной дискриминации, то есть даже серии с приемлемым профилем поведения in vivo могут быть отбракованы с помощью такого метода контроля качества.
Исследование биодоступности
31. Необходимо представить краткое описание исследований биодоступности, которое включает в себя информацию о:
а) фармакокинетике (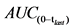 ,
, 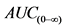 Cmax) и в соответствующих случаях другие значимые фармакокинетические параметры (Cmin в равновесном состоянии, частичную AUC, отношение Cmax/Cmin и т.д.);
Cmax) и в соответствующих случаях другие значимые фармакокинетические параметры (Cmin в равновесном состоянии, частичную AUC, отношение Cmax/Cmin и т.д.);
б) точечной оценке и 90-процентных доверительных интервалах фармакокинетических параметров для воспроизведенных лекарственных препаратов;
в) производственных площадках и датах производства;
г) номерах и размерах серии;
д) составах и результатах растворения использованных серий.
32. Исследования биодоступности необходимо проводить с сериями лекарственного препарата размером 100 000 единиц или не менее 10% от размера серии полномасштабного производства, в зависимости от того, какой из данных показателей больше, если только с сериями такого размера не проводились опорные клинические исследования. При этом достаточно провести исследования биодоступности, используя серии лекарственного препарата меньшего размера, если данные серии были произведены способом, соответствующим полномасштабному производственному процессу. Например, если клинические исследования II фазы (включая фармакокинетические исследования и исследования биодоступности) проводятся на серии с массой 15 килограммов, при этом уже проведены опорные клинические испытания на серии с массой 60 килограммов (при этом исследования биодоступности данной серии отсутствуют), и предполагается, что будет полномасштабное производство серии в 600 килограммов, то проведение дополнительных исследований биодоступности для серии в 60 килограммов не требуется.
Сравнение профилей растворения
33. В ряде случаев для установления подобия должны быть сопоставлены профили растворения, например:
после масштабирования, изменения состава и (или) производственного процесса;
в случае экстраполяции результатов in vivo при регистрации различных дозировок.
Подобие профилей растворения должно быть установлено с использованием не менее 12 индивидуальных значений для временной точки. Необходимо учесть временные точки и частоту отбора проб, учитывая физико-химические in vitro и in vivo характеристики действующего вещества и механизм высвобождения лекарственного препарата.
34. В случае экстраполяции результатов in vivo при регистрации различных дозировок лекарственного препарата (в отсутствие in vivo сравнительных данных по всем дозировкам исследуемого препарата с препаратом сравнения) растворение других дозировок исследуемого препарата следует сравнить с дозировкой испытуемого препарата, использованной в исследовании биоэквивалентности.
35. Следует сравнить профили растворения. При этом установление их подобия также может требовать подтверждения статистическими методами с использованием модельно-независимых или модельно-зависимых критериев, таких как:
а) оценка линейной регрессии доли (количества в процентах) действующего вещества, растворившегося в определенных временных точках;
б) статистическое сравнение параметров функции Вейбулла;
в) расчет коэффициента подобия;
г) другие модельно-независимые или модельно-зависимые критерии (при обосновании).
Установление корреляции in vivo - in vitro
36. Испытание на растворение in vitro является важным в целях обеспечения необходимого постоянства качества от серии к серии и является показателем постоянства в пределах серии (когда все единицы лекарственной формы имеют желаемые функциональные характеристики in vivo). Путем установления четкой корреляции между характеристиками высвобождения in vitro и параметрами биодоступности in vivo испытание на растворение in vitro может служить в качестве суррогатного маркера поведения in vivo, а также показателем постоянства терапевтических свойств рутинно производимых серий лекарственного препарата. При установлении корреляции следует регистрировать и анализировать вариабельность данных. Как правило, чем выше вариабельность данных, используемых для разработки корреляции in vivo - in vitro, тем меньше уверенность в оценках параметров модели и тем выше неопределенность в ее прогнозировании поведения in vivo.
37. Установленная корреляция in vivo - in vitro уровня A позволяет сократить число исследований in vivo в процессе разработки лекарственного препарата, используется в составлении спецификаций и содействует принятию определенных регуляторных решений (например, масштабирование и внесение пострегистрационных изменений). В этой связи заявитель должен рассмотреть возможность разработки такой корреляции in vivo - in vitro. Кроме того, установление корреляции in vivo - in vitro уровня A позволяет уверенно использовать испытание на растворение как инструмент управления изменениями. В качестве альтернативы для сравнения in vitro и in vivo данных допускается использовать механистическую модель (например, фармакокинетические модели, основанные на физиологии (PBPK)).
38. Валидация корреляции in vivo - in vitro уровня A заключается в подтверждении достаточности ее прогностической способности. Корреляция in vivo - in vitro уровня A устанавливается, например, на основе методики деконволюции, с помощью которой абсорбцию in vivo или растворение in vivo можно спрогнозировать, исходя из данных исследования in vitro (Указания по установлению вида корреляции изложены в приложении к настоящему Руководству). Валидированная корреляция in vivo - in vitro уровня A позволяет использовать связанное с ней испытание на растворение in vitro в качестве суррогатного маркера для исследований in vivo, поскольку полученный профиль зависимости "концентрация in vivo - время" можно спрогнозировать с использованием результатов испытания на растворение in vitro и уравнения корреляции in vivo - in vitro. Данный подход предполагает следующее:
такая корреляция in vivo - in vitro уровня C может надежно использоваться только для интерполяции;
одна модель корреляции in vivo - in vitro должна применяться ко всем составам лекарственной формы, использованным при разработке и валидации модели;
корреляция in vivo - in vitro не может служить основанием признания биоэквивалентности лекарственных препаратов от разных заявителей на основании исключительно данных in vitro.
39. Модель корреляции in vivo - in vitro следует применять в целях интерполяции в пределах диапазона данных, использованных для ее разработки, а не в целях экстраполяции за пределами ее рабочего диапазона. Указанный принцип исследования особенно важен при подаче заявлений в регуляторные органы (например, обоснование спецификации растворения и в случае биовейвера), что имеет определяющее значение при выборе составов лекарственной формы, включенных в исследование корреляции in vivo - in vitro.
40. Для разработки и валидации корреляции in vivo - in vitro рекомендуется, как правило, использовать составы с широко варьирующими профилями растворения in vitro, поскольку использование составов с небольшими различиями в их профилях растворения in vitro ограничит возможности для расширения диапазона спецификации и диапазона, в пределах которого может быть обоснован биовейвер. Следует учитывать, что в случае крайних вариантов составов могут вступать в действие различные механизмы высвобождения и другие биофармацевтические факторы, влияя на зависимость высвобождения действующего вещества в условиях in vitro и in vivo и препятствуя получению одного уравнения корреляции in vivo - in vitro, которое описывало бы поведение всех составов в пределах диапазона, предложенного для биовейвера. Исходя из этого, составы должны быть выбраны таким образом, чтобы тот же самый механизм (по возможности) контролировал бы высвобождение действующего вещества как in vitro, так и in vivo. Как правило, это ограничивает диапазон профилей растворения in vitro, используемых на практике для разработки и валидации корреляции in vivo - in vitro.
41. Если для дальнейшей разработки корреляции in vivo - in vitro впоследствии выбран крайний состав (т.е. состав с самым быстрым или с самым медленным растворением in vitro из числа составов, использованных в корреляции in vivo - in vitro), целесообразно расширить диапазон валидации корреляции in vivo - in vitro путем получения данных в условиях in vivo для другого состава (с более быстрым или медленным растворением в зависимости от обстоятельств) и использования этих данных для внешней валидации существующей корреляции in vivo - in vitro или повторной разработки и валидации новой корреляции in vivo - in vitro. Таким образом, важно, чтобы планируемый целевой состав был должным образом окружен крайними вариантами.
2. Составление спецификаций
42. Спецификация составляется с использованием дискриминирующего испытания на растворение.
43. Как правило, в спецификацию на растворение in vitro лекарственного препарата с пролонгированным высвобождением для приема внутрь включается не менее 3 точек:
а) ранняя временная точка для исключения демпинга дозы и (или) установления характеристик нагрузочной (начальной) дозы (обычно от 20% до 30% растворенного вещества);
б) не менее одной точки для обеспечения соответствия форме профиля растворения (около 50% растворенного вещества);
в) одна точка для обеспечения высвобождения большей части действующего вещества (Q = 80%). Если максимальное количество растворенного вещества составляет менее 80%, то последней временной точкой должно быть время достижения профилем растворения своего плато.
44. Для лекарственных препаратов с нулевым порядком высвобождения спецификация скорости (времени) растворения для заданного интервала времени может быть более подходящая, чем суммарное количество растворенного вещества в отдельной временной точке. Если кинетика высвобождения нулевого порядка сочетается с переменным лаг-периодом (время задержки эффекта), такая спецификация является обязательной. Методика установления лаг-периода определяется заявителем.
45. Приемлемую вариацию, допустимую вокруг каждой временной точки (верхнюю и нижнюю границы), можно установить различными способами:
а) в отсутствии корреляции in vivo - in vitro. Допустимые пределы могут быть получены на основании разброса данных о растворении in vitro серий с подтвержденными приемлемыми функциональными характеристиками in vivo (биосерия (биосерии)) или путем доказательства биоэквивалентности серий в предложенных верхней и нижней границах диапазона растворения (концепция "крайней серии"). Обычно допустимый диапазон значений высвобождения в любой заданный момент времени не должен превышать общую численную разницу на ![]() 10% от заявленного содержания действующего вещества, то есть общая вариабельность должна составлять 20% (например, при заявленном содержании действующего вещества 50
10% от заявленного содержания действующего вещества, то есть общая вариабельность должна составлять 20% (например, при заявленном содержании действующего вещества 50 ![]() 10% это обозначает, что допустимый диапазон составляет от 40% до 60%), если более широкий диапазон не подтвержден исследованиями биоэквивалентности;
10% это обозначает, что допустимый диапазон составляет от 40% до 60%), если более широкий диапазон не подтвержден исследованиями биоэквивалентности;
б) установленная корреляция in vivo - in vitro уровня A. Валидированная корреляция in vivo - in vitro уровня A позволяет использовать данные растворения in vitro (предложенные, а не полученные в ходе наблюдения данные) для замены исследования in vivo составов в предложенных пределах спецификации на растворение. Профили растворения получают из предложенных пределов с помощью установленной корреляции in vivo - in vitro, предпочтительно включающей соответствующее математическое описание характеристик функции растворения in vitro (функции Вейбулла, уравнения Хилла и т.д., основанные на поведении испытуемых составов при разработке лекарственного препарата), или, что менее предпочтительно, основываются на высвобождении в различных временных точках. Полный профиль зависимости "концентрация в плазме - время" рассчитывается для предложенных верхнего и нижнего пределов растворения, а также с учетом полученных данных растворения in vitro для состава, планируемого для регистрации (состава сравнения), с использованием валидированной корреляции in vivo - in vitro. Соответствующую Cmax и выбранные значения параметра AUC рассчитывают для предложенных нижнего и верхнего пределов, состава сравнения и полученных отношений (верхнего предела к нижнему, верхнего предела к пределу для состава сравнения и нижнего предела к пределу для состава сравнения).
46. Основной принцип составления спецификации заключается в том, что все серии с показателями между нижним и верхним пределами спецификации на растворение должны быть биоэквивалентными друг к другу. Если биоэквивалентность основана на данных in vivo, допустимый диапазон для максимальной разницы по результатам сравнения составляет от 80,00% до 125,00%, основанный на доверительных интервалах вокруг средних значений Cmax и выбранного параметра AUC. Несмотря на то, что некоторые методики анализа корреляции in vivo - in vitro позволяют количественно определять биологическую вариабельность и прогнозировать доверительные интервалы, большинство методик прогнозируют лишь средние данные зависимости "концентрация - время". Таким образом, критерии установления границ признания биоэквивалентности, прогнозируемой на основе средних значений (с использованием данных растворения вместо данных in vivo и подтвержденных корреляций in vivo - in vitro), должны быть жестче, то есть разница в значениях Cmax и выбранного параметра AUC для средних значений зависимости "концентрация - время" в испытании in vivo, прогнозируемых для верхнего и нижнего пределов спецификации на растворение, должна быть менее 20%. Границы, основанные на разнице, превышающей 20% между прогнозируемыми значениями Cmax и выбранным параметром AUC для верхнего и нижнего пределов спецификации на растворение, должны быть обоснованы производителем лекарственного препарата.
47. AUC лекарственных препаратов, всасывающихся на всем протяжении желудочно-кишечного тракта, между различными составами с широко варьирующими скоростями растворения зачастую схожа, поэтому спецификация составляется на основании показателя Cmax, а не показателя AUC. Преимущество использования корреляции in vivo - in vitro для составления спецификации в данном случае заключается в том, что в определенные временные точки границы кумулятивного растворения могут выходить за пределы ![]() 10%, поскольку величина влияния различных временных точек на показатель Cmax неодинаковая. Чувствительность показателя Cmax к изменениям в растворении зависит от фармакокинетических свойств (чем короче период полувыведения, тем больше чувствительность к изменениям в растворении) и формы зависимости корреляции in vivo - in vitro (в зависимости от того, что происходит быстрее: растворение лекарственного препарата in vitro или in vivo).
10%, поскольку величина влияния различных временных точек на показатель Cmax неодинаковая. Чувствительность показателя Cmax к изменениям в растворении зависит от фармакокинетических свойств (чем короче период полувыведения, тем больше чувствительность к изменениям в растворении) и формы зависимости корреляции in vivo - in vitro (в зависимости от того, что происходит быстрее: растворение лекарственного препарата in vitro или in vivo).
3. Стратегия контроля качества
48. Общие требования по разработке и обоснованию стратегии контроля качества лекарственного препарата предусматриваются соответствующими актами органов Союза. Следует осуществлять контроль критических показателей качества, необходимых для контроля высвобождения лекарственного средства.
49. В ходе фармацевтической разработки следует установить связь (качественную или количественную) фармакокинетических параметров посредством высвобождения in vivo действующего вещества со скоростью растворения in vitro.
50. Во время углубленной фармацевтической разработки соответствие лекарственной формы лекарственного препарата требованиям испытания на растворение in vitro может подтверждаться с помощью испытаний при выпуске лекарственного препарата в режиме реального времени. Поскольку скорость высвобождения действующего вещества может быть чувствительна к масштабированию, необходимо, чтобы методика прогнозирования скорости высвобождения действующего вещества была верифицирована в условиях полномасштабного производства.
4. Внесение изменений в регистрационное досье
лекарственного препарата
51. Требования к данным, обосновывающим внесение изменений в регистрационное досье, зависят от степени значимости изменения, наличия корреляции in vivo - in vitro уровня A, наличия или необходимости изменения методики (пределов) растворения. Если данные биодоступности (биоэквивалентности) не представлены, необходимо обосновать их отсутствие.
52. Если корреляция in vivo - in vitro уровня A установлена, а спецификация высвобождения не меняется, изменения могут быть приняты на основании данных in vitro, терапевтического индекса действующего вещества и прогностической способности корреляции in vivo - in vitro. В этом случае отказ производителя лекарственного препарата от необходимости проведения исследований биоэквивалентности должен основываться на сопоставлении прогнозируемых профилей зависимости "концентрация в плазме - время" и связанных с ними фармакокинетических параметров для составов до изменений и после них, рассчитанных с использованием данных in vitro и валидированной корреляции in vivo - in vitro.
53. В отношении лекарственных препаратов с доказанной корреляцией уровня B или C или в отсутствии корреляции in vivo - in vitro необходимо представить данные о биодоступности (биоэквивалентности), если не представлено обоснование отсутствия таких данных.
V. Лекарственные формы с отложенным
(отсроченным) высвобождением
1. Основные положения
54. Фармакопея Союза определяет несколько лекарственных форм с отложенным (отсроченным) высвобождением: кишечнорастворимые капсулы, таблетки и гранулы. В данном разделе представлены специальные указания для кишечнорастворимых лекарственных форм. Лекарственные препараты, основанные на других принципах, также часто классифицируются как лекарственные формы с отложенным (отсроченным) высвобождением, в том числе разработанные для высвобождения в определенном участке желудочно-кишечного тракта под влиянием определенного триггер-фактора (например, ферменты) или в определенное время после приема внутрь. Несмотря на то, что описанные в настоящем Руководстве принципы фармацевтической разработки, составления спецификаций и стратегии контроля качества применимы также и к другим лекарственным формам с отложенным (отсроченным) высвобождением, для таких лекарственных форм целесообразно разработать отдельное руководство на основе принципа подбора соответствующего состава и механизма высвобождения.
55. Многие из положений, приведенные для лекарственных форм с пролонгированным высвобождением для приема внутрь, применимы также для лекарственных форм с отложенным (отсроченным) высвобождением.
2. Фармацевтическая разработка
56. Резюме исследований биоэквивалентности, которое необходимо представить в регистрационном досье, включает в себя информацию о фармакокинетике (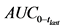 ,
, 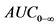 , Cmax и, если приемлемо, другие параметры (например, частичную AUC, для воспроизведенных лекарственных препаратов - также точечную оценку и 90-процентные доверительные интервалы), производственных площадках и датах производства, номерах и размерах серии, составах лекарственной формы и результатах растворения использованных серий.
, Cmax и, если приемлемо, другие параметры (например, частичную AUC, для воспроизведенных лекарственных препаратов - также точечную оценку и 90-процентные доверительные интервалы), производственных площадках и датах производства, номерах и размерах серии, составах лекарственной формы и результатах растворения использованных серий.
57. Следует указать цель отложенного (отсроченного) высвобождения, например, защита слизистой оболочки желудка, защита действующего вещества от воздействия кислой среды желудка или целенаправленное высвобождение действующего вещества в заданном сегменте желудочно-кишечного тракта для оказания местного действия и т.д.
58. Следует проанализировать механизм высвобождения и обосновать выбор вспомогательного вещества (вспомогательных веществ), ответственного за отложенное (отсроченное) высвобождение, например, целенаправленное высвобождение при заданном значении рН, чувствительность к воздействию ферментов, эрозии со временем и т.д.
В ходе фармацевтической разработки следует установить качественную или количественную связь, характеризующую высвобождение in vivo, между фармакокинетическими параметрами и скоростью растворения in vitro.
В зависимости от поведения лекарственного препарата в желудке можно выделить 2 вида составов препаратов с отложенным (отсроченным) высвобождением:
одноединичные (цельные) нераспадающиеся лекарственные формы;
распадающиеся лекарственные формы, содержащие гранулы.
59. Не рекомендуется, как правило, осуществлять разработку одноединичных (цельных) нераспадающихся кишечнорастворимых лекарственных форм гастрорезистентных лекарственных препаратов, поскольку период времени их нахождения в желудке непредсказуем и больше, чем у распадающихся лекарственных форм, содержащих гранулы. Поэтому такие одноединичные (цельные) нераспадающиеся кишечнорастворимые лекарственные формы подвержены более высокому риску демпинга дозы и (или) имеют беспорядочные профили концентрации.
60. Если общая характеристика лекарственного препарата требует одновременного приема с пищей или не исключает его, то испытание на гастрорезистентность следует проводить также в условиях, характерных для состояния сытости. Например, для определения устойчивости при высвобождении в полном желудке, испытания должны проводиться при более высоком значении pH (например, в диапазоне 3,0 - 5,0) с использованием как нераспадающихся цельных лекарственных форм, так и распадающихся лекарственных форм, содержащих гранулы. Большое количество пищи в желудке временно приводит к повышению pH до значения 3,0 или выше, поэтому испытание при pH со значением 2,0 не будет являться в достаточной степени подтверждающим.
3. Составление спецификаций
61. В спецификацию на растворение in vitro кишечнорастворимого лекарственного препарата должно быть включено не менее 2 временных точек:
а) ранняя временная точка для исключения высвобождения в кислой среде (менее 10% растворенного вещества через 2 часа);
б) одна точка для обеспечения высвобождения основного количества действующего вещества в нейтральной или близкой к нейтральной среде.
62. Гастрорезистентность должна подтверждаться в течение 2 часов или более.
63. Критерии приемлемости для последующего этапа испытания указаны в Фармакопее Союза.
4. Стратегия контроля качества
64. Требования к разработке и обоснованию стратегии контроля качества лекарственного препарата приводятся в соответствующих нормативных актах, входящих в право Союза. Необходимо осуществлять контроль критических показателей качества, ответственных за отсроченное (отложенное) высвобождение лекарственного средства (например, целостность кишечнорастворимой оболочки).
65. В ходе проведения фармацевтической разработки следует установить качественную или количественную связь, характеризующую высвобождение in vivo, между фармакокинетическими параметрами и скоростью растворения in vitro. В условиях углубленной фармацевтической разработки соответствие требованиям растворения может подтверждаться посредством испытаний при выпуске лекарственного препарата в режиме реального времени. Поскольку скорость высвобождения лекарственных форм с отсроченным (отложенным) высвобождением может быть чувствительна к масштабированию, необходимо верифицировать проектное поле в условиях полномасштабного производства.
5. Внесение изменений в регистрационное досье
лекарственного препарата
66. Поскольку испытание на растворение in vitro гастрорезистентных лекарственных форм с отсроченным (отложенным) высвобождением считается релевантным для условий испытаний in vivo, изменение вспомогательных веществ, ответственных за отсроченное (отложенное) высвобождение лекарственных препаратов в гастрорезистентной лекарственной форме, допускается обосновать исключительно на основе полученных результатов in vitro испытаний (при наличии обоснований). Профили высвобождения, полученные в результате испытаний на гастрорезистентность, должны быть неизменными.
Приложение
к Руководству по качеству лекарственных
препаратов с модифицированным
высвобождением для приема внутрь
УКАЗАНИЯ ПО УСТАНОВЛЕНИЮ ВИДА КОРРЕЛЯЦИИ
I. Корреляция in vivo - in vitro
1. Для установления уровня корреляции in vivo - in vitro используются различные методы. Различают следующие уровни корреляции in vivo - in vitro:
а) уровень A отражает поточечную зависимость между кривой растворения препарата in vitro и кривыми растворения in vivo, полученными методом деконволюции данных о концентрации в плазме (метод Вагнера-Нельсона, Лу-Ригельмана, численная деконволюция) или другими соответствующими методами (методы моделирования, основанные на конволюции или дифференциальных уравнениях с использованием средних данных, или методы моделирования популяционной фармакокинетики);
б) уровень B отражает одноточечную зависимость по одному из показателей:
между средним временем растворения лекарственного препарата in vitro и средним временем удерживания in vivo или средним временем растворения in vivo с использованием принципов анализа статистических моментов;
между константой скорости растворения in vitro (kd) и полученной константой скорости абсорбции (kabs);
в) уровень C отражает одноточечную зависимость между количеством вещества, растворенного in vitro за определенное время, и средним значением одного из фармакокинетических параметров (например, AUC, Cmax или Tmax). Если один или несколько фармакокинетических параметров коррелирует с количеством растворенного действующего вещества в нескольких временных точках профиля растворения, считается установленной множественная корреляция уровня C.
II. Разработка корреляции in vivo - in vitro
Уровень A
2. Рекомендации по дизайну исследования и последующему анализу данных корреляции in vivo - in vitro изложены в актах, входящих в право Евразийского экономического союза. Как правило, в процессе перекрестных исследований, проводимых у здоровых добровольцев, применяются 2 или более составов с достаточно различающимися профилями растворения и соответствующий состав сравнения (для целей деконволюции) с быстрым высвобождением действующего вещества (например, раствор для внутривенного введения, раствор для приема внутрь или лекарственная форма с немедленным высвобождением). Концентрация исходного (неизмененного) действующего вещества в крови или плазме определяется как функция времени. Корреляцию in vivo - in vitro можно моделировать непосредственно по концентрации действующего вещества в плазме (одноэтапный подход) или после деконволюции профилей "концентрация - время" для состава с модифицированным высвобождением относительно состава с немедленным высвобождением (двухэтапный подход). Чтобы испытание на растворение in vitro служило суррогатным маркером поведения in vivo и использовалось в качестве инструмента контроля изменений, как правило, требуется корреляция in vivo - in vitro уровня A.
3. Первоначальное изучение составов в различных испытаниях (условиях) на растворение при выпуске лекарственного препарата позволяет установить испытание, обеспечивающее наиболее подходящую дискриминационную способность. Временные точки отбора проб в испытании на растворение in vitro для составов, используемых в исследовании корреляции in vivo - in vitro, должны быть достаточно частыми, чтобы полностью охарактеризовать профиль растворения, в том числе плато (например, 3 последовательные точки, различающиеся не менее чем на 5%). Меньшее число временных точек для отбора проб допускается устанавливать при проведении испытания контроля качества, однако обратное не является справедливым: временные точки для отбора проб при проведении контроля качества не применимы в качестве точек для испытания на растворение in vitro с целью получения данных корреляции in vivo - in vitro, поскольку разреженные данные могут не позволить осуществить точную интерполяцию между точками, и прекращение отбора проб до достижения плато приводит к неполному высвобождению и нарушает валидацию корреляции in vivo - in vitro.
Уровни B и C
4. Как правило, корреляция уровней B и C неприменима для обоснования производителем значимых изменений состава или процесса производства лекарственного препарата. Однако множественная корреляция уровня C может служить в качестве вспомогательного средства в составлении спецификации.
5. Разработка множественной корреляции уровня C проводится путем установления линейной корреляции на основе не менее 3 временных точек между количеством растворенного вещества в 3 или более временных точках или 3 точках MDT, с одной стороны, и соответствующими показателями AUC и Cmax для ряда составов с различными профилями скорости растворения in vitro, показателем MRT или любым другим подходящим фармакокинетическим параметром (множественная корреляция уровня C), с другой стороны. Данные in vitro могут использоваться для прогнозирования функциональных характеристик in vivo. Следует отметить, что если множественная корреляция уровня C достижима, то также возможна разработка корреляции уровня A. Корреляция in vivo - in vitro уровня A позволяет прогнозировать полный профиль зависимости "концентрация в плазме - время" (предоставляя необходимую информацию о форме профиля и времени достижения максимальной концентрации) в дополнение к общим фармакокинетическим параметрам, таким как Cmax и AUC, в то время как из множественной корреляции уровня C можно прогнозировать лишь обобщающие фармакокинетические параметры. Следовательно, корреляция уровня A является предпочтительным подходом.
6. Параметры, применяемые для установления корреляции in vivo - in vitro различных уровней, приведены в таблице.
Таблица
Параметры, применяемые для установления корреляции
in vivo - in vitro различных уровней
Уровень
Вид зависимости
Параметр
in vitro
in vivo
A
поточечная
профиль растворения
фармакокинетическая кривая
B
одноточечная
MDTvitro
kd
MRT, MDTvivo
kabs
C
одноточечная
rd (T20-30%, T50%, T80%)
Cmax Tmax, AUC (средние значения)
Множественная корреляция
C (T20-30%, T50%, T80%)
или C (MDT)
Cmax, Tmax, AUC, MRT и др.
III. Оценка прогностической способности корреляции
in vivo - in vitro
7. При использовании корреляции in vivo - in vitro в качестве суррогатного маркера функциональных характеристик in vivo следует верифицировать, что прогнозирование функциональных характеристик in vivo, основанное на профиле растворения in vitro, применимо для скоростей растворения in vitro, охватываемых корреляцией in vivo - in vitro. Такая оценка должна быть сведена к оценке прогнозирования функциональных характеристик или, наоборот, погрешности прогнозирования.
8. При оценке прогностической способности особое значение имеет учет 2 основных аспектов:
а) чем меньше данных, доступных для разработки и оценки корреляции in vivo - in vitro, тем больше дополнительных данных требуется для полной оценки прогностической способности корреляции in vivo - in vitro;
б) исследуемые составы должны надлежащим образом отличаться по скорости высвобождения (например, не менее чем на 10% от растворенного количества), что приводит к значительному различию рассматриваемых фармакокинетических параметров.
9. Методология и отчет об анализе прогностической способности применяются в соответствии с руководством по фармакокинетическому и клиническому изучению биоэквивалентности лекарственных препаратов с модифицированным высвобождением, биоэквивалентности липосомальных препаратов, биоэквивалентности кортикостероидов для местного применения в дерматологии, утверждаемым Евразийской экономической комиссией.